
Willkommen bei der Zeitschrift für Infektionstherapie!
INFEKTIO aktuell
Aktuelle Informationen zur Infektionstherapie
++ Up to date bleiben: Newsletter-Anmeldung ++
Ab Januar 2025 – Die neue App für Ihr Digital-Abo

Alle neuen Ausgabe der Zeitschrift für Infektionstherapie finden Sie ab 2025 nur noch auf der neuen digitalen Abo-Plattform – als App oder im Browser!
Die Vorteile:
- Responsives Design
- Tag- und Nacht-Lesemodus
- Vorlesefunktion
- SPECIAL: Fragen Sie den Wissens-Chat, das integrierte KI-Tool, nach Suchbegriffen und fachlichen Antworten!
Hier geht's...
Fragen? Schauen Sie in unsere FAQ-Liste.
Cefazolin vs. Antistaphylokokken-Penicilline bei Methicillin-sensibler Staphylococcus aureus-Bakteriämie
Die Bakteriämie durch Staphylococcus aureus zählt weltweit zu den führenden bakteriell bedingten Todesursachen; mehr als jeder vierte Betroffene verstirbt innerhalb von 90 Tagen. Die Wahl des optimalen Antibiotikums bei Methicillin-sensibler S. aureus (MSSA)-Bakteriämie bleibt umstritten. Endokarditis-Leitlinien favorisieren traditionell die Antistaphylokokken-Penicilline (Nafcillin, Oxacillin, Cloxacillin oder Flucloxacillin) gegenüber Cefazolin – vor allem aufgrund des sogenannten Cefazolin-Inokulumeffekts, einer In-vitro-Zerstörung von Cefazolin durch β-Lactamasen bei hoher Erregerlast. Meta-Analysen von Beobachtungsstudien deuten hingegen auf eine mögliche Überlegenheit von Cefazolin hinsichtlich der 30-Tage-Sterblichkeit hin (Odds Ratio 0,73; 95-%-Konfidenzintervall [KI] 0,62 bis 0,85).
Im Rahmen der internationalen Bayesianischen adaptiven Plattformstudie „Staphylococcus aureus Network Adaptive Platform“ (SNAP) wurde zwischen dem 17. Februar 2022 und dem 7. August 2024 an
91 Zentren in acht Ländern ein offener, randomisierter Vergleich von Cefazolin mit Flucloxacillin oder Cloxacillin bei erwachsenen Patienten mit Penicillin-resistenter MSSA-Bakteriämie durchgeführt.
Empfohlen wurden 2 g Cefazolin i.v. alle 8 Stunden (bzw. alle 6 Stunden bei schwerer Erkrankung oder Endokarditis), 2 g Flucloxacillin i.v. alle 6 Stunden (bzw. alle 4 Stunden) oder 2 g Cloxacillin
i.v. alle 4 Stunden. Primärer Endpunkt war die Gesamtsterblichkeit innerhalb von 90 Tagen; die Nichtunterlegenheitsgrenze wurde als adjustierte Odds Ratio von < 1,2 definiert (entsprechend einem
absoluten Mortalitätsunterschied von < 2,5 Prozentpunkten bei 15 % Sterblichkeit in der Vergleichsgruppe).
Von 1.341 randomisierten Patienten (671 Cefazolin, 670 Antistaphylokokken-Penicillin; medianes Alter 66 Jahre [Interquartilsbereich 53 bis 76]; 31,4 % Frauen) gingen 1.287 in die Analyse des primären
Endpunkts ein. Häufigster Infektionsherd war eine osteoartikuläre Infektion (32,1 %). Hinsichtlich des primären Endpunkts erfüllte Cefazolin das Kriterium der Nichtunterlegenheit gegenüber
Flucloxacillin oder Cloxacillin mit einer posterioren Wahrscheinlichkeit von 99,2 %; die Wahrscheinlichkeit einer Überlegenheit lag bei 89,8 %. Auch die Sterblichkeit an den Tagen 14, 28 und 42 sowie
das Auftreten einer akuten Nierenschädigung, definiert als absoluter Kreatininanstieg um mindestens 26,5 µmol/l (0,3 mg/dl) innerhalb von 5 Tagen oder relativer Anstieg um mindestens 50 % gegenüber
dem Ausgangswert innerhalb der ersten 14 Tage, fielen zugunsten von Cefazolin aus. Ebenso traten schwerwiegende, mit dem Studienmedikament assoziierte unerwünschte Reaktionen sowie Therapieabbrüche
aufgrund unerwünschter Ereignisse unter Cefazolin deutlich seltener auf (Tab. 1).
Tab. 1: Primäre und sekundäre Endpunkte im Vergleich Cefazolin vs. Flucloxacillin/Cloxacillin
| Endpunkt | Cefazolin | Flucloxacillin/ Cloxacillin | Adjustierte Odds Ratio (95-%-Kredibilitätsintervall) |
| Sterblichkeit bis Tag 90 (primärer Endpunkt) | 97/645 (15,0 %) | 109/642 (17,0 %) | 0,81 (0,59 bis 1,12) |
| Sterblichkeit bis Tag 14 | 3,7 % | 5,6 % | 0,67 (0,39 bis 1,11) |
| Sterblichkeit bis Tag 28 | 7,1 % | 10,5 % | 0,61 (0,41 bis 0,91) |
| Sterblichkeit bis Tag 42 | 9,5 % | 13,0 % | 0,66 (0,46 bis 0,94) |
| Akute Nierenschädigung innerhalb 14 Tage | 92/660 (13,9 %) | 127/648 (19,6 %) | 0,67 (0,50 bis 0,89) |
| Schwerwiegende unerwünschte Reaktionen | 12/671 (1,8 %) | 33/670 (4,9 %) | 0,41 (0,21 bis 0,77) |
| Therapieabbruch wegen unerwünschter Ereignisse | 11/671 (1,6 %) | 61/670 (9,1 %) | 0,21 (0,11 bis 0,38) |
Folgerung der Autoren
Bei Erwachsenen mit Methicillin-sensibler S. aureus-Bakteriämie war Cefazolin gegenüber Flucloxacillin oder Cloxacillin hinsichtlich der 90-Tage-Gesamtsterblichkeit nicht unterlegen und mit
einer geringeren Inzidenz akuter Nierenschädigungen sowie weniger schwerwiegenden unerwünschten Ereignissen assoziiert. Bedenken hinsichtlich der klinischen Relevanz des Cefazolin-Inokulumeffekts
sollten durch die enge Nichtunterlegenheitsgrenze sowie das mit der Gesamtpopulation vergleichbare Therapieergebnis bei Patienten mit Endokarditis wesentlich relativiert werden.
Kommentar der Herausgeber
Diese wichtige und lang ersehnte Studie legt einen Paradigmenwechsel in der Therapie der S. aureus-Bakteriämie nahe, was sich in künftigen Leitlinien widerspiegeln dürfte. Nicht nur finden
sich weniger unerwünschte Nebenwirkungen mit Cefazolin, sondern auch eine niedrigere Gesamtmortalität. In aller Regel ist nun eine Therapie mit Cefazolin vorzuziehen.
The Staphylococcus aureus Network Adaptive Platform (SNAP) Trial Group. Cefazolin for Methicillin-Susceptible Staphylococcus aureus Bacteremia. N Engl J Med. 2026; 394(23):2329-2339. doi: 10.1056/NEJMoa2506905
15.07.2026
Ebola-Ausbruch durch Bundibugyo-Virus in der Demokratischen Republik Kongo und Uganda weitet sich aus
Der seit Mai 2026 anhaltende Ausbruch der Bundibugyo-Virus-Erkrankung (BVD) in der Demokratischen Republik Kongo (DRK) und Uganda breitet sich weiter aus. Nach Angaben der WHO wurden bis Anfang Juli 2026 insgesamt 1.481 bestätigte Fälle gemeldet, davon 1.460 aus der DRK, 20 aus Uganda sowie ein Fall aus Frankreich (mit Bezug zur DRK). Bislang sind 454 Todesfälle registriert worden, während sich 229 Patient:innen bereits erholt haben.
Die BVD ist eine schwere Form der Ebola-Erkrankung, verursacht durch eine Spezies des Genus Orthoebolavirus, wobei Flughunde als vermutetes natürliches Reservoir gelten. Die Übertragung von Mensch zu
Mensch erfolgt durch direkten Kontakt mit Blut, Sekreten oder anderen Körperflüssigkeiten infizierter Personen sowie mit kontaminierten Oberflächen. Nach einer Inkubationszeit von 2 bis 21 Tagen
treten die ersten Symptome auf. Frühsymptome wie Fieber, Müdigkeit, Muskel- und Kopfschmerzen sowie Halsschmerzen sind unspezifisch und erschweren die klinische Diagnose. Im weiteren Verlauf treten
gastrointestinale Symptome, Organdysfunktion und in einigen Fällen hämorrhagische Manifestationen auf. Die CFR der beiden vorangegangenen BVD-Ausbrüche in Uganda (2007) und der DRK (2012) betrugen 30
% bzw. 50 %.
In der DRK betrifft der aktuelle Ausbruch 36 Gesundheitszonen der Provinzen Ituri, Nord-Kivu und Süd-Kivu mit einer rohen Fallsterblichkeitsrate von 30,9 %. Unter medizinischem Personal wurden 102
bestätigte Fälle einschließlich 25 Todesfälle gemeldet. Der Ausbruch verläuft in einem komplexen humanitären und konfliktgeprägten Umfeld mit hoher Bevölkerungsmobilität, was die Ausbruchskontrolle
zusätzlich erschwert. In Uganda traten die 20 bestätigten Fälle bislang ausschließlich in den Distrikten Kampala und Wakiso auf; eine Übertragung in der Bevölkerung wurde nicht dokumentiert. In
Frankreich wurde am 24. Juni 2026 ein laborbestätigter Fall bei einem Mediziner nach fünfwöchigem Einsatz in Ituri gemeldet. Der Patient wurde am Flughafen isoliert und in eine
Hochsicherheits-Behandlungseinrichtung verlegt.
Da bislang keine zugelassenen Impfstoffe oder spezifischen Therapien gegen BVD existieren, stützt sich die Ausbruchskontrolle auf rasche Fallidentifikation, Isolierung, Kontaktnachverfolgung, sichere
Bestattungen und intensives Community Engagement. Die WHO stuft das Risiko in der DRK als sehr hoch, in Uganda sowie in den angrenzenden Ländern als hoch ein. Für die übrige Region Afrika und global
wird das Risiko als niedrig bewertet. Reise- oder Handelsbeschränkungen werden derzeit nicht empfohlen.
15.07.2026
INFEKTIO_Podcast | Oberflächenprobleme – Infektiologische Aspekte in der Dermatologie
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuelle Folge ist Prof. Mario Fabri aus Jena zu Gast. Gemeinsam widmen sie sich den infektiologischen Aspekten der Dermatologie.
15.07.2026
Begrenzte Budgets, viele Impfstoffe: Welche Programme im Gavi-Portfolio bringen den größten Nutzen?
Angesichts wachsender Finanzierungsengpässe in der globalen Gesundheitsversorgung und einer stetig wachsenden Zahl verfügbarer Impfstoffe gewinnt die Frage an Bedeutung, welche Impfprogramme den größten gesundheitlichen Nutzen pro investierter Dosis erbringen. Die vorliegende Modellierungsstudie des Vaccine Impact Modelling Consortium hatte zum Ziel, Impf-Effekt-Verhältnisse (vaccine impact ratios) – definiert als verhinderte Todesfälle bzw. behinderungsbereinigte Lebensjahre (disability-adjusted life-years, DALYs) pro 1000 Impfungen – für 14 von Gavi, the Vaccine Alliance, unterstützte Impfprogramme in 117 Niedrig- und Mitteleinkommensländern (LMICs) zu schätzen und damit eine standardisierte Vergleichsgrundlage zu schaffen.
Berücksichtigt wurden neue Schätzungen für COVID-19, Malaria und Cholera sowie aktualisierte Schätzungen für Hepatitis B (HepB), humanes Papillomavirus (HPV), Masern, Meningitis (MenA ab 2010 mit
Umstellung auf MenACWYX ab 2027), Röteln, Typhus und Gelbfieber, ergänzt durch bestehende Schätzungen für Japanische Enzephalitis, Haemophilus influenzae Typ b (Hib), Rotaviren (Rota) und
Pneumokokken-Konjugatimpfstoff (PCV). Die Modellierungsgruppen verwendeten standardisierte demografische Inputs und harmonisierte Impfquoten-Annahmen einschließlich eines kontrafaktischen Szenarios
ohne Impfung. Pro Erkrankung wurden in der Regel mindestens zwei Modelle einbezogen, um strukturelle, parameter- und stochastische Unsicherheit abzubilden. Der Zeithorizont reichte von 2000 bis 2030,
für Cholera bis 2040; die Krankheitslast wurde bis 2100 modelliert, um die Lebensspanne der geimpften Kohorten zu berücksichtigen.
In der Gesamtbetrachtung über alle Impfaktivitäten und Subregionen hinweg wiesen HPV und Masern die höchsten Impf-Effekt-Verhältnisse hinsichtlich verhinderter Todesfälle und DALYs auf. Die mittleren
Impf-Effekt-Verhältnisse für alle eingeschlossenen Impfstoffe sind in der folgenden Tabelle dargestellt:
| Impfstoff | Verhinderte Todesfälle pro 1000 Impfungen (95-%-KI) | Verhinderte DALYs pro 1000 Impfungen (95-%-KI) |
| HPV |
11,24 (10,88 bis 11,64) |
523,04 (499,92 bis 547,11) |
| Masern | 6,09 (4,90 bis 7,07) | 411,01 (331,63 bis 476,14) |
| HepB |
5,00 (4,47 bis 5,58) |
141,94 (127,28 bis 157,71) |
| Malaria (RTS,S/R21) | 2,78 (2,16 bis 3,53) | 203,04 (156,99 bis 259,97) |
| Hib | 2,22 (1,81 bis 2,61) | 150,37 (124,74 bis 176,12) |
| Gelbfieber | 1,86 (0,52 bis 4,23) | 72,26 (20,06 bis 165,63) |
| Rota | 0,80 (0,49 bis 1,04) | 46,07 (25,78 bis 61,78) |
| Röteln | 0,22 (0,12 bis 0,35) | 16,78 (11,91 bis 23,51) |
| Typhus | 0,68 (0,21 bis 1,72) | 26,57 (7,96 bis 70,69) |
| PCV | 1,53 (0,87 bis 2,09) | 104,91 (61,47 bis 142,93) |
| Meningokokken-Meningitis (MenA/MenACWYX) | 0,66 (0,49 bis 0,85) | 42,78 (31,90 bis 54,91) |
| Cholera | 0,20 (0,13 bis 0,56) | 6,13 (4,81 bis 12,61) |
| Japanische Enzephalitis | 0,18 (0,10 bis 0,30) | 80,57 (55,55 bis 106,80) |
| COVID-19 | 0,12 (0,08 bis 0,18) | 4,03 (2,86 bis 6,19) |
Aufgrund der parameter-, struktur- und stochastikbedingten Unsicherheit überlappten die Wertebereiche häufig. Bemerkenswert sind Unterschiede in der Rangfolge zwischen verhinderten Todesfällen und DALYs: So rangiert Malaria, die überwiegend sehr junge Kinder betrifft und zu vielen verlorenen Lebensjahren führt, bei den DALYs höher als bei den Todesfällen. Die Impf-Effekt-Verhältnisse variierten zudem zwischen den Subregionen sowie nach Impfaktivitätstyp (Routineimmunisierung vs. Kampagne) und Dosis. Für die HPV-Impfung beziehen sich die Ergebnisse auf ein Zwei-Dosen-Schema; der Übergang vieler Länder zu einem Ein-Dosen-Schema würde den Effekt pro Dosis deutlich verbessern. Beim Vergleich von MenA und MenACWYX zeigte MenACWYX trotz überlappender Unsicherheitsbereiche tendenziell ein höheres Impf-Effekt-Verhältnis bei verhinderten Todesfällen. Die Röteln-Schätzungen umfassen ausschließlich Todesfälle durch das kongenitale Rötelnsyndrom und nicht Infektionen während der Schwangerschaft mit möglichen Aborten oder Totgeburten.
Folgerung der Autoren
Mit der Einführung länderspezifischer Impfstoffbudgets im Rahmen von Gavi 6.0 (2025–30) gewinnt eine rigorose Priorisierung der Impfprogramme an Bedeutung, da die verfügbaren Mittel nicht alle empfohlenen Impfstoffe abdecken können. Die berechneten Impf-Effekt-Verhältnisse bieten hierfür eine standardisierte und reproduzierbare Vergleichsgrundlage. HPV- und Masernimpfungen weisen die höchsten Werte für verhinderte Todesfälle und DALYs pro 1000 Impfungen auf; für andere Impfstoffe variieren die Verhältnisse nach Subregion, Aktivitätstyp und Dosis, mit häufig überlappenden Unsicherheitsbereichen. Die Ergebnisse sollten daher gemeinsam mit weiteren Faktoren wie Kosteneffektivität und Einsatz in der Ausbruchsbekämpfung betrachtet werden, um Impfinvestitionen evidenzbasiert zu optimieren.
Kommentar der Herausgeber
In der individuellen Impfberatung ist eine Priorisierung von einzelnen Impfungen gegenüber anderen heikel und häufig kontraproduktiv. Für die öffentliche Gesundheit und die Entscheidung, welche
Impfprogramme angesichts limitierter Ressourcen besonders wirksam sind, haben diese Zahlen dagegen eine große Bedeutung.
Gaythorpe, K. A. M. et al. Quantifying relative health impact across Gavi, the Vaccine Alliance’s portfolio in 117 countries at the subregional level: a modelling study. Lancet. 2026; 407(10542):1941-1952. doi: 10.1016/S0140-6736(26)00000-0
17.06.2026
Mundpflege als wirksame Prävention der nicht-beatmungsassoziierten nosokomialen Pneumonie (NV-HAP)
Die nicht-beatmungsassoziierte nosokomiale Pneumonie (non-ventilator hospital-acquired pneumonia, NV-HAP) zählt zu den häufigsten nosokomialen Infektionen, wird jedoch nur selten systematisch erfasst oder gezielt durch Präventionsmaßnahmen adressiert. Patienten mit NV-HAP verbringen zwischen 10 und 48 Tage länger im Krankenhaus und haben ein etwa achtfach erhöhtes Sterberisiko. Die Mundpflege gilt als wesentliche präventive Maßnahme, da eine Reduktion der oralen bakteriellen Belastung das Risiko aspirationsbedingter Pneumonien senken kann. Bisher liegen jedoch nur wenige hochwertige randomisierte kontrollierte Studien aus dem Krankenhaussetting vor.
Die Hospital Acquired Pneumonia Prevention (HAPPEN)-Studie war eine offene, multizentrische, stepped-wedge, cluster-randomisierte kontrollierte Studie an drei australischen Krankenhäusern. Neun Clustern wurden drei Behandlungssequenzen zugeordnet. Eingeschlossen waren alle Patienten ab 18 Jahren mit einer Verweildauer von mindestens 48 h. Die Intervention umfasste drei Komponenten:
- intensivierte Bereitstellung von Mundpflege und entsprechenden Produkten
- Patienten- und Mitarbeiterschulungen
- Audit und Feedback
Die Mundpflege beinhaltete das Befeuchten der Lippen sowie die Reinigung von Zähnen oder Zahnprothesen und Zunge mit einer geeigneten Zahnbürste und einer natriumbikarbonat- und fluoridhaltigen Zahnpasta; antiseptische Mundspülungen kamen nicht zum Einsatz. Die Kontrollbedingung bestand aus der üblichen Praxis. Der primäre Endpunkt war die NV-HAP-Inzidenz nach den Kriterien des European Centre for Disease Prevention and Control (ECDC). Sekundäre Endpunkte umfassten nosokomiale Infektionen der unteren und oberen Atemwege (LRTI bzw. URTI, exklusive COVID-19) sowie Infektionen der Mundhöhle (EENT-Oral).
Im Studienzeitraum vom 3. Juni 2024 bis zum 22. August 2025 waren 12.446 Patienten teilnahmeberechtigt; 3.336 wurden wegen einer Verweildauer von unter 48 Stunden ausgeschlossen. In die Analyse gingen 8.870 Patienten ein, davon 4.523 unter Kontroll- und 4.347 unter Interventionsbedingung. Eine NV-HAP trat bei insgesamt 78 (0,9 %) Patienten auf, davon 46 (1,0 %) von 4.523 in der Kontroll- und 32 (0,7 %) von 4.347 in der Interventionsgruppe. Die Interventionsexposition war mit einer kumulativen Hazard Ratio von 0,40 (95-%-KI: 0,19 bis 0,82) verbunden, was einer Reduktion von 1,00 auf 0,41 Infektionen pro 100 Aufnahmetage unter Risiko entsprach. Für nosokomiale Infektionen der unteren oder oberen Atemwege lag die kumulative Hazard Ratio bei 1,64 (95-%-KI: 0,61 bis 4,39), für Infektionen der Mundhöhle bei 1,08 (95-%-KI: 0,66 bis 1,77). Der Anteil der Patienten mit vollständig umgesetztem Mundpflegeprotokoll erhöhte sich unter der Intervention deutlich von 474 (15,9 %) von 2.988 auf 1727 (61,9 %) von 2791.
Folgerung der Autoren
Diese multizentrische, cluster-randomisierte Studie zeigt, dass eine Strategie zur Verbesserung der Mundpflege die NV-HAP-Inzidenz bei hospitalisierten Patienten im Vergleich zur Standardversorgung wirksam reduziert. Für nosokomiale Infektionen der unteren oder oberen Atemwege sowie der Mundhöhle wurde kein Nutzen nachgewiesen, was möglicherweise damit zusammenhängt, dass die primären Übertragungswege viraler Infektionen durch Mundpflege nicht beeinflusst werden. Über die NV-HAP-Prävention hinaus dürften weitere Vorteile in der Linderung von Mundtrockenheit, der Verbesserung des Patientenkomforts sowie der früheren Erkennung von Komplikationen wie oraler Candidiasis liegen. Die Ergebnisse stärken die Evidenzbasis für zukünftige Leitlinien zur NV-HAP-Prävention. Da die Intervention durch dedizierte Forschungspflegekräfte erbracht wurde, sind weitere implementierungsorientierte Studien sowie gesundheitsökonomische Analysen erforderlich.
Kommentar der Herausgeber
Während zahlreiche aktuelle Studien in der Infektionsprävention verschiedene Bundle von oft zahlreichen Maßnahmen testen, zeigte diese Arbeit den Nutzen einer relativen einfachen intensivierten oralen Hygienemaßnahme.
White, N. M. et al. Effectiveness of oral care for the prevention of non-ventilator hospital-acquired pneumonia (HAPPEN): a multicentre, stepped-wedge, cluster-randomised trial in Australia. Lancet Infect Dis. 2026; published online first June 8, 2026. doi: 10.1016/S1473-3099(26)00235-5
17.06.2026
INFEKTIO_Podcast | Ich hab da was gefunden – Fallstricke der mikrobiologischen Diagnostik
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuelle Folge ist Prof. Holger Rohde aus Hamburg zu Gast. Gemeinsam widmen sie sich den Fallstricken der mikrobiologischen Diagnostik.
17.06.2026
Trends bei der Langzeit-Antibiotikaverordnung nach gezielten ambulanten Antibiotic-Stewardship-Maßnahmen
Bis zu 90 % des Antibiotikaverbrauchs entfallen auf den ambulanten Bereich, wobei schätzungsweise die Hälfte aller Verordnungen suboptimal oder unnötig ist. Übermäßig lange Therapiedauern finden
sich je nach Syndrom in 40–85 % der Verordnungen und tragen maßgeblich zur Entwicklung von Antibiotikaresistenzen bei. Die Mayo Clinic implementierte im Januar 2020 ein ambulantes
Antibiotic-Stewardship-Programm (ASP), das sich auf Primärversorgung und Akutversorgung (Urgent Care) konzentrierte und jährlich syndromspezifische Verbesserungsziele verfolgte – etwa die Reduktion
unnötiger Antibiotikaverordnungen bei viralen oberen Atemwegsinfektionen (URI) und die Optimierung der Verordnungspraxis bei Harnwegsinfektionen (UTI). Eine retrospektive, multizentrische
Kohortenstudie untersuchte nun, ob diese fokussierten Maßnahmen auch mit breiteren Reduktionen von Langzeit-Antibiotikaverordnungen (LDAP; definiert als > 7 Tage) korrespondierten.
Insgesamt wurden 675.526 Antibiotikaverordnungen aus 642.579 Patientenkontakten im Zeitraum November 2020 bis Juni 2025 eingeschlossen; 42,2 % wiesen eine Verordnungsdauer > 7 Tage auf.
ASP-Einrichtungen machten 83,0 % aller Kontakte aus. ASP-Verordnungspanels wurden in 10,5 % aller Kontakte genutzt, wobei 99,2 % der Nutzung auf ASP-Einrichtungen entfielen. Bei Panel-Nutzung lag die
LDAP-Rate bei 30,1 % gegenüber 44,2 % ohne.
Abbildung 1 zeigt den Gesamttrend der LDAP über den Studienzeitraum von November 2020 bis Juni 2025, dargestellt als monatlicher Anteil aller ambulanten Kontakte mit einer Antibiotikaverordnung >
7 Tage. Der Anteil sank von 46,2 % im November 2020 auf 35,7 % im Juni 2025, was einer absoluten Differenz von −10,5 Prozentpunkten (pp) bzw. einer relativen Reduktion von 22,7 % entspricht. Die
lineare Regressionslinie modelliert eine geschätzte jährliche Abnahme von −3,2 pp. Auffällig ist ein vorübergehender Anstieg der LDAP von etwa November 2022 bis Juni 2023, der mit der Rückkehr zu
einer „normalen“ respiratorischen Virussaison in den USA nach der COVID-19-Pandemie korrespondiert.
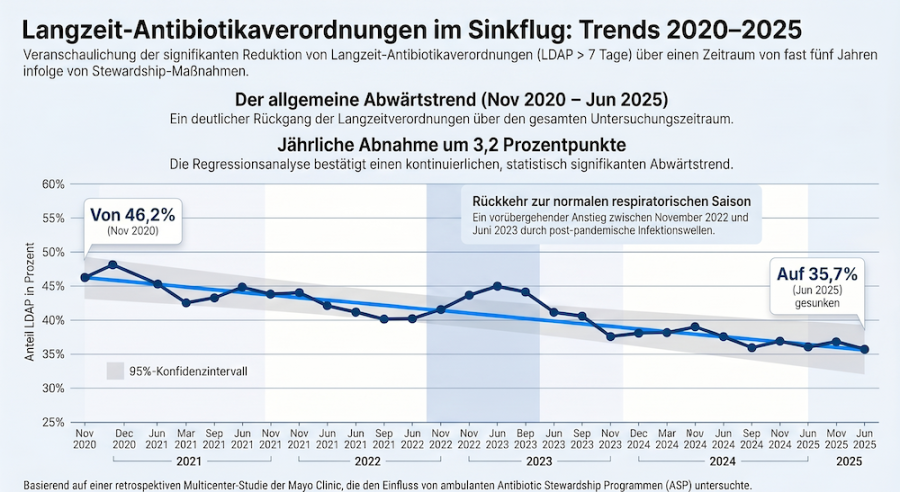
Abbildung 1: Gesamtanteil ambulanter Kontakte mit Antibiotikaverordnungen > 7 Tage über die Zeit (November 2020 – Juni 2025). Die blaue Trendlinie modelliert die geschätzte jährliche Abnahme von −3,2 Prozentpunkten; der grau schattierte Bereich zeigt das zugehörige 95-%-Konfidenzintervall.
Eigene Darstellung basierend auf Daten aus: [1], erstellt mit NotebookLM
Die Subgruppenanalysen zeigten, dass sowohl ASP- als auch Nicht-ASP-Einrichtungen signifikante Rückgänge der LDAP über den Studienzeitraum verzeichneten, wobei die Verbesserungen in
ASP-Einrichtungen deutlich ausgeprägter waren. Die LDAP in ASP-Einrichtungen sank von 46,8 % auf 34,8 % (Differenz −12 pp), während Nicht-ASP-Einrichtungen einen Rückgang von 45,5 % auf 39,1 %
verzeichneten (Differenz −6,4 pp). In der linearen Regression zeigten Nicht-ASP-Einrichtungen eine signifikante jährliche Abnahme von 2,4 pp (95-%-Konfidenzintervall [KI] 1,8 bis 2,9; P < 0,001).
Für ASP-Einrichtungen betrug die jährliche Abnahme 3,4 pp (95-%-KI 2,9 bis 3,9; P < 0,001) – 1,0 pp mehr als in Nicht-ASP-Einrichtungen (95-%-KI 0,26 bis 1,8; P = 0,009). Bei ausschließlicher
Betrachtung der Nicht-Prioritätsdiagnosen war die Abnahme in ASP-Einrichtungen mit 4,1 pp pro Jahr (95-%-KI 3,8 bis 4,5; P < 0,001) signifikant größer als in Nicht-ASP-Einrichtungen mit 2,7 pp
(95-%-KI 2,3 bis 3,0; P < 0,001), mit einer Differenz von 1,4 pp (95-%-KI 0,9 bis 1,9; P < 0,001). Innerhalb der ASP-Einrichtungen wiesen Nicht-Prioritätsdiagnosen eine jährliche Abnahme von
4,1 pp auf (95-%-KI 3,3 bis 4,9; P < 0,001), während Prioritätsdiagnosen eine Abnahme von 2,6 pp zeigten (95-%-KI 1,9 bis 3,4; P < 0,001), wobei die Differenz 1,5 pp betrug (95-%-KI 0,4 bis
2,6; P = 0,010). Innerhalb Nicht-ASP-Einrichtungen nahmen Nicht-Prioritätsdiagnosen um 2,7 pp pro Jahr ab (95-%-KI 2,0 bis 3,4; P < 0,001), während Prioritätsdiagnosen mit einer geschätzten
jährlichen Veränderung von −0,4 pp stabil blieben (95-%-KI −1,1 bis 0,3; P = 0,281). Die Differenz zwischen den beiden Gruppen betrug 2,3 pp (95-%-KI 1,3 bis 3,3; P < 0,001).
Bemerkenswert war, dass in ASP-Einrichtungen auch bei Nicht-Prioritätsdiagnosen – also Syndromen, die nie direkt vom ASP adressiert wurden – signifikante LDAP-Reduktionen auftraten. In
Nicht-ASP-Einrichtungen hingegen blieben Prioritätsdiagnosen hinsichtlich der LDAP stabil. Respiratorische Diagnosen außerhalb der URI-Prioritätsgruppe zeigten mit 7,4 pp pro Jahr (95-%-KI 6,6 bis
8,2; P < 0,001) die größte jährliche Abnahme.
Folgerung der Autoren
Die Befunde deuten auf vorteilhafte „Off-Target“-Effekte hin: Obwohl die Interventionen syndromspezifisch ausgerichtet waren, korrelierte die Präsenz des ASP in ASP-Einrichtungen mit Verbesserungen
der Verordnungspraxis auch bei nie adressierten Syndromen. Klinische Entscheidungsunterstützungsinstrumente wie Verordnungspanels mit standardisierten Therapiedauern ≤ 7 Tage trugen wahrscheinlich
wesentlich zur LDAP-Reduktion bei. Die LDAP erweist sich als praktikable Benchmark-Metrik für ambulante ASPs. Weitere Studien sind erforderlich, um diese Ergebnisse zu bestätigen.
Kommentar der Herausgeber
Die meisten Antibiotika werden im ambulanten Sektor verordnet. Diese Arbeit zeigt, dass auch hier mit ASP sehr viel erreicht und ein Kulturwandel bewirkt werden kann. Jetzt müssen wir nur noch
entsprechende Strukturen aufbauen und fördern.
[1] Ilges, D. et al. Trends in Long Duration Antibiotic Prescribing Following Focused Outpatient Antimicrobial Stewardship Efforts Across a Large Healthcare Enterprise. Clin Infect Dis. 2025; published online first April 09, 2026. doi: 10.1093/cid/ciag244
06.05.2026
Sterblichkeitstrends bei Patienten mit schwer behandelbaren, antibiotikaresistenten Gram-negativen Infektionen im Zeitalter neuer Antibiotika in den USA
Gram-negative Infektionen mit schwer behandelbarer Resistenz (Difficult-to-treat Resistance, DTR) sind definiert als Resistenz gegenüber sämtlichen First-Line-Antibiotika, d.h. allen β-Lactamen
einschließlich Carbapenemen sowie Fluorchinolonen, und weisen eine um 40 % höhere Sterblichkeit auf als empfindliche Infektionen. Historisch erforderten DTR-Infektionen den Einsatz von Antibiotika
wie Aminoglykosiden, Polymyxinen oder Tigecyclin, die mit suboptimalen Behandlungsergebnissen assoziiert sind. Seit 2014 wurden in den USA mehrere neue Antibiotika mit Aktivität gegen resistente
Gram-negative Erreger zugelassen, darunter Ceftazidim-Avibactam, Ceftolozan-Tazobactam, Meropenem-Vaborbactam, Imipenem-Relebactam, Cefiderocol und Eravacyclin. Ob diese Substanzen tatsächlich zu
verbesserten Überlebensraten bei Patienten mit DTR-Infektionen geführt haben, war bislang unklar.
Eine retrospektive Kohortenstudie untersuchte anhand der PINC-AI Healthcare Database, ob und warum sich die Sterblichkeit bei Patienten mit DTR-Gram-negativen Infektionen seit Einführung dieser
neueren Antibiotika verändert hat. Eingeschlossen wurden erwachsene stationäre Patienten (Alter ≥ 18 Jahre), die zwischen dem 1. Januar 2016 und dem 31. August 2023 entlassen wurden und bei denen ein
mikrobiologischer Nachweis einer DTR-Infektion durch Enterobacterales, Pseudomonas aeruginosa oder Acinetobacter baumannii vorlag, verbunden mit einer Antibiotikatherapie über
mindestens drei konsekutive Tage. Im Studienzeitraum wurden 8 319 398 stationäre Aufenthalte aus 471 Krankenhäusern erfasst, von denen 9384 (0,11 %) einen DTR-Erregernachweis aufwiesen. Davon
erfüllten 5065 (54,0 %) Aufenthalte aus 262 Krankenhäusern die Einschlusskriterien. Das mittlere Alter betrug 59,9 Jahre (Standardabweichung 16,2), 59,5 % waren männlich. Eine Sepsisdiagnose lag bei
64,8 % vor, 45,7 % erforderten eine Intensivaufnahme, 31,7 % eine maschinelle Beatmung und 24,8 % Vasopressoren. Die häufigsten Erreger waren P. aeruginosa (46,6 %), A. baumannii
(28,6 %) und Enterobacterales (24,8 %).
Die Verfügbarkeit neuerer Antibiotika nahm im Studienzeitraum deutlich zu: Der Anteil der Krankenhäuser mit Einsatz mindestens eines neueren Antibiotikums stieg von 12 % (23 von 198) im Zeitraum
2016–2018 auf 63 % (103 von 163) im Zeitraum 2021–2023; der Anteil mit Empfindlichkeitstestungen für neuere Antibiotika von 4 % (8 von 198) auf 71 % (116 von 163). Der Anteil der Patienten, die ein
neueres Antibiotikum als Initialtherapie erhielten, stieg von 4 % (21 von 589) im Jahr 2016 auf 15 % (34 von 234) im Jahr 2023. Dennoch erhielten in der Mehrzahl der Fälle (196 [84 %] von 234) die
Patienten auch 2023 weiterhin eine in-vitro-diskordante initiale Antibiotikatherapie. Insgesamt war bei 3775 von 4875 Aufenthalten (77,4 %) die Initialtherapie inadäquat. Von den 1100 Aufenthalten
mit adäquater Therapie wurde in 783 Fällen (71,2 %) dennoch ein traditionelles DTR-aktives Antibiotikum eingesetzt.
Die Krankenhaussterblichkeit wurde mittels eines generalisierten linearen gemischten Modells (GLMM) analysiert, adjustiert für patienten-, krankenhaus- und COVID-19-pandemiebezogene Faktoren. Über
den gesamten Zeitraum gemittelt betrug die geschätzte Sterblichkeitswahrscheinlichkeit 20 % (95-%-Konfidenzintervall [KI] 18 bis 23) für Enterobacterales, 20 % (95-%-KI 18 bis 22) für P.
aeruginosa und 25 % (95-%-KI 23 bis 28) für A. baumannii. Zwischen 2016 und 2023 wurde keine Veränderung des durchschnittlichen marginalen Effekts auf die adjustierte Sterblichkeit
beobachtet – weder für Enterobacterales (0,1 %; 95-%-KI −1,1 bis 1,4) noch für P. aeruginosa (−0,7 %; 95-%-KI −1,7 bis 0,3) oder A. baumannii (−0,4 %; 95-%-KI −1,8 bis 0,9).
Bei Stratifizierung nach Infektionsort zeigte sich einzig für DTR-P. aeruginosa-Blutstrominfektionen eine Abnahme der adjustierten Sterblichkeit (−4,5 % pro Jahr; 95-%-KI −8,2 bis −0,6),
während alle anderen Erreger-Infektionsort-Kombinationen unverändert blieben. Im GLMM war weder der Erhalt eines neueren Antibiotikums (adjustierte Odds Ratio [aOR] 1,11; 95-%-KI 0,89 bis 1,40) noch
eine in-vitro-diskordante vs. konkordante Initialtherapie (aOR 1,12; 95-%-KI 0,91 bis 1,38) mit einer veränderten Sterblichkeit assoziiert. Auch die Verfügbarkeit neuerer Antibiotika auf
Krankenhausebene zeigte keinen signifikanten Zusammenhang mit der Sterblichkeit.
Folgerung der Autoren
Trotz erheblich gestiegener Verfügbarkeit und Verordnung neuerer Antibiotika in US-amerikanischen Krankenhäusern zwischen 2016 und 2023 wurde bei der Mehrzahl der DTR-Gram-negativen Infektionen keine
Reduktion der Sterblichkeit beobachtet. Die initiale Antibiotikatherapie bleibt überwiegend diskordant; selbst bei konkordanter Therapie werden häufig suboptimale traditionelle DTR-aktive Substanzen
eingesetzt. Die konventionelle Empfindlichkeitstestung benötigt durchschnittlich 92,2 Stunden, was eine erhebliche Verzögerung der zielgerichteten Therapie bedeutet. Die Entwicklung und
Implementierung schneller diagnostischer Plattformen zur frühzeitigen Erkennung resistenter Phänotypen, klinische Studien mit stärkerer Repräsentation von DTR-Patienten sowie die Erforschung
nicht-antimikrobieller, wirtgerichteter Therapieansätze könnten entscheidende Schritte zur Senkung der Sterblichkeit bei resistenten Infektionen darstellen.
Kommentar der Herausgeber
Obwohl die neueren, deutlich teureren Antibiotika in Zulassungsstudien nahezu durchweg mit einem besseren Outcome assoziiert sind als ältere Substanzen, zeigen die enttäuschenden Ergebnisse aus der
Praxis – begründet in der offensichtlichen Bevorzugung billigerer alter Substanzen und der zu langsamen Diagnostik und des dadurch verzögerten Einsatzes – deutlich, dass der durch Studien belegte
Vorteil dieser neuen Mittel nur durch einen entsprechend gut gesteuerten Einsatz in einen klinischen Vorteil für die Patienten überführt werden kann.
Walker, M. K. et al. Survival trends in patients with difficult-to-treat, antibiotic-resistant, Gram-negative infections in the era of next-generation antibiotics in the USA: a retrospective cohort study. Lancet Infect Dis. 2026; published online first March 25, 2026. doi: 10.1016/S1473-3099(26)00020-4
06.05.2026
Neue S3-Leitlinie zur Tuberkuloseprävention bei neu zugewanderten Menschen in Deutschland
Tuberkulose ist in Deutschland mit einer Inzidenz von etwa
5 / 100.000 Einwohnerinnen und Einwohnern selten, doch rund 75 % der Betroffenen sind nicht in Deutschland geboren. Für 2022 wurden 4.076 Fälle gemeldet, 71 % der 116 Todesfälle betrafen Menschen mit nicht-deutscher Staatsangehörigkeit. Die Inzidenz für eine extrapulmonale Tuberkulose ist 64-fach höher bei ausländischer vs. deutscher Staatsangehörigkeit.
Das Deutsche Zentralkomitee zur Bekämpfung der Tuberkulose (DZK) hat unter Beteiligung von 16 Fachgesellschaften nun die erste deutsche S3-Leitlinie zur Tuberkuloseprävention bei neu zugewanderten Menschen vorgelegt (AWMF-Register 020-029, Version 1.0, September 2025).
Die Leitlinie umfasst 31 konsensbasierte Empfehlungen für das Screening auf Tuberkuloseinfektion (TBI), die präventive Therapie, das Screening auf aktive Tuberkulose, die Implementierungsvoraussetzungen sowie Kommunikationsstrategien.
Dabei verwendet sie ein dreistufiges Empfehlungsschema: „soll" (starke Empfehlung) bedeutet, dass Evidenz und Nutzen-Risiko-Abwägung klar sind; „sollte" (schwache Empfehlung) signalisiert eingeschränkte Evidenz, bei der individuelle Faktoren berücksichtigt werden sollten; „kann" (offene Empfehlung) wird bei unsicherer Evidenzlage ausgesprochen.
TBI-Screening – altersabhängig abgestuft:
- 15–35 Jahre, Herkunftsland-Inzidenz > 100 / 100.000, Einreise innerhalb der letzten 2 Jahre: soll angeboten werden (starker Konsens 100 %)
- 35–45 Jahre, gleiche Kriterien: sollte angeboten werden (Konsens 95 %)
- > 45 Jahre, Inzidenzschwellenwert > 150 / 100.000: kann nach individueller Nutzen-Risiko-Abwägung angeboten werden (Konsens 82 %)
- Komplexe Migrationsbedingungen (z.B. lange Fluchtrouten, Kriegs-/Krisenregionen): kann unabhängig von Alter und Inzidenz angeboten werden (starker Konsens 100 %)
Die abgeschwächte Empfehlung für über 45-Jährige begründet sich durch die ansteigende Rate toxisch bedingter Hepatitiden unter Isoniazid und das abnehmende Lebenszeitprogressionsrisiko.
Populationsbasierte Kohortenstudien zeigten eine Reduktion der Tuberkulose-Inzidenz durch Screening und präventive Therapie (HR 0,76; 95-%-Konfidenzintervall [KI] 0,63 bis 0,91). Das kumulative
2-Jahres-Progressionsrisiko ohne Therapie beträgt 4,1 % (95-%-KI 1,3 bis 12,0), wobei 81,6 % des 5-Jahres-Risikos in den ersten 2 Jahren auftreten.
Präventive Therapie – bevorzugte Regime:
- 4R (Rifampicin 4 Monate): gleiche Wirksamkeit wie 9H, höhere Abschlussraten (+157 / 1.000), weniger UAW Grad 3–5
- 3HR (Isoniazid + Rifampicin 3 Monate): Effektivität vs. Placebo OR 0,53 (95-%-KI 0,36 bis 0,78), weniger UAW vs. 6-9H, höhere Abschlussraten (+389 / 1.000)
- 6-9H nur bei Kontraindikation/Unverträglichkeit von Rifampicin
- Bei MDR-/RR-Tuberkulose-Rate > 10 % im Herkunftsland (v.a. postsowjetische Staaten): individuelle Abwägung mit spezialisiertem Zentrum
- Kontaktpersonen bei Fluorchinolon-empfindlichem MDR-/RR-Stamm: Levofloxacin 6 Monate (Risikoreduktion: Kinder 56 %, Erwachsene/Jugendliche 45 %)
Tuberkulose-Screening:
- Ab 15 Jahren, Herkunftsland-Inzidenz > 100 / 100.000, geplanter Aufenthalt > 3 Monate: TBI-Screening sollte zeitnah nach Einreise angeboten werden
- Bei zusätzlichen Risikofaktoren oder in Settings mit Ausbruchspotential oder in vulnerablen Gruppem: TBI-Screening soll angeboten werden bei Herkunftsland-Inzidenz > 100 / 100.000 oder MDR-/RR-Prävalenz > 10%
- Methode: Kombination Symptom-Screening + Thoraxröntgen
- Entdeckung > 84 % der prävalenten Fälle;
- Fallfindungsrate bei Asylsuchenden in Deutschland: 347 / 100.000, NNS: 288
Kinder und Jugendliche (< 15 Jahre):
- TBI-Screening bei Herkunft aus Ländern mit Inzidenz > 100 / 100.000 (TBI-Rate durchschnittlich 11,1 %; bei unbegleiteten minderjährigen Geflüchteten 18,0 %)
- Geflüchtete Kinder < 5 Jahre: immer Screening, auch ohne weitere Risikofaktoren
Implementierung:
- Screening nach Einreise, kostenfrei, freiwillig, unabhängig vom Aufenthaltsstatus
- Fallmanagement über die gesamte Versorgungskaskade
- Monitoring und Evaluation hinsichtlich Kosteneffektivität und epidemiologischem Impact
Kommunikationsstrategien:
- Schulung von Fachpersonal zu Tuberkulose, Versorgungskette sowie kultursensibler und diskriminierungsfreier Kommunikation
- Aufklärung zur Entkräftung kultureller Fehleinschätzungen und Vermittlung zentraler Botschaften (Tuberkulose ist heilbar; klare Trennung von Screening und Aufenthaltsrecht)
- Zielgruppenorientierte, sprach- und kultursensible Kommunikation unter Einbezug von Sprachmittlung oder Übersetzungshilfen
Die Leitliniengruppe betont, dass die erfolgreiche Umsetzung aller Empfehlungen eine kostendeckende Finanzierung sämtlicher Screeningkomponenten sowie eine bundesweite, systematische Evaluation der
Versorgungskaskade voraussetzt, um die Maßnahmen evidenzbasiert weiterzuentwickeln und langfristig zur Tuberkulose-Elimination in Deutschland beizutragen.
Häcker, B. et al. S3-Leitlinie Tuberkuloseprävention bei neu zugewanderten Menschen (TB-Risk); AWMF-Register Nr. 020-029. 2025; Version 1.0, September 2025. URL: https://register.awmf.org/de/leitlinien/detail/020-029
08.04.2026
Neue Surviving Sepsis Campaign 2026: Was Infektiologen und ABS-Experten wissen müssen
Die SSC-Leitlinie ist die weltweit meistzitierte und einflussreichste Leitlinie zum Management der Sepsis – getragen von der Society of Critical Care Medicine (SCCM) und der European Society of Intensive Care Medicine (ESICM), mit 69 Panelisten aus 23 Ländern. Die aktuelle Version 2026 umfasst 129 Empfehlungen und Statements und adressiert erstmals Themen wie prähospitale Antibiotikagabe, Betablocker-Einsatz und selektive Darmdekontamination.
Die SSC 2026 enthält einige neue Empfehlungen, die für Antibiotic Stewardship (ABS) und infektiologische Beratung unmittelbar relevant sind.
Anaerobier-wirksame Therapie
Bei Patienten ohne Risikofaktoren für anaerobe Infektionen – insbesondere bei pulmonalem oder urologischem Fokus – soll keine routinemäßige anaerobier-wirksame Therapie erfolgen (bedingte Empfehlung).
Selektive Darmdekontamination (SDD)
Erstmals empfiehlt die SSC 2026 SDD bei beatmeten Patienten – jedoch ausdrücklich nur in Einrichtungen mit niedriger MRE-Prävalenz (bedingte Empfehlung). Die S3-Leitlinie 2025 enthält hierzu keine Empfehlung. Für Deutschland ist die Indikation an lokalen Resistenzstatistiken zu orientieren.
Procalcitonin
PCT wird von der SSC 2026 zur Steuerung der Therapiedauer empfohlen (bedingte Empfehlung), zur Einleitung der Antibiotikatherapie hingegen nicht. Die S3-Leitlinie spricht zur Therapiedauer eine starke Empfehlung aus.
Fungale Biomarker
Sowohl Beta-D-Glucan als auch Mannan werden von der SSC 2026 nicht zur Einleitung oder Beendigung einer empirischen Antimykotikatherapie empfohlen. Die S3-Leitlinie nennt Beta-D-Glucan als einen von mehreren Risikofaktoren, ohne eine biomarkergesteuerte Strategie formal zu empfehlen.
Prescott, H. C. et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock 2026. Intensive Care Med. 2026; published online first. doi: 10.1007/s00134-026-08361-1
08.04.2026
Zulassungsempfehlung für ersten Kombinationsimpfstoff gegen COVID-19 und Influenza
Nachdem erst kürzlich der neue mRNA-Impfstoff gegen COVID-19 mNEXSPIKE (Moderna) in Europa zugelassen wurde, könnte dieser bald auch in Kombination mit einem Grippeimpfstoff zur Verfügung stehen [1]. Der Ausschuss für Humanarzneimittel (CHMP) bei der europäischen Arzneimittelbehörde EMA empfiehlt nach seiner Sitzung am 26. Februar die Zulassungserteilung für mCOMBRIAX, den ersten kombinierten Impfstoff gegen COVID-19 und saisonale Influenza für Menschen ab 50 Jahren [2].
Die durch COVID-19 und Influenza verursachten Atemwegsinfektionen verlaufen in den meisten Fällen leicht bis mittelschwer, bei älteren Menschen und Personen mit geschwächtem Immunsystem können jedoch
schwere Verläufe auftreten, insbesondere bei einer Koinfektion mit Influenza und SARS-CoV-2 [3]. In Deutschland wurden seit Beginn der Saison 2025/2026 etwa 225.000 Influenza-Fälle (26 %
hospitalisiert) und 111.000 COVID-19-Fälle (36 % hospitalisiert) gemäß Infektionsschutzgesetz an das Robert Koch-Institut gemeldet [4]. Unter den ca. 1.500 bzw. 1.400 Todesfällen mit bestätigter
Influenza- bzw. SARS-CoV-2 Infektion waren 95 % bzw. 96 % 60 Jahre oder älter.
mCOMBRIAX (mRNA-1083) ist eine Kombination aus mNEXSPIKE (mRNA-1283), dem gegen SARS-CoV2-gerichteten mRNA-Impfstoff der zweiten Generation, und mRNA-1010, dem gegen die Influenza-Viren A/H1N1,
A/H3N2 und B/Victoria gerichteten Grippeimpfstoff [2,5].
mCOMBRIAX enthält pro Einzeldosis insgesamt 40 µg mRNA, verkapselt in Lipid-Nanopartikeln [5]. Im Gegensatz zu allen vorherigen mRNA-basierten Impfstoffen gegen COVID-19 codiert die in mNEXSPIKE
enthaltenen mRNA nicht für das volle Spike-Protein, sondern nur für die membrangebundene N-terminale Domäne und die rezeptorbindende Domäne [2, 5]. Die in mRNA-1010 enthaltenen mRNA-Sequenzen
codieren hingegen für die vollständigen membrangebundenen Hämagglutinin-Glykoproteine der saisonalen Influenzaviren [2,5].
Die Zulassungsempfehlung basiert auf den Ergebnissen einer randomisierten, beobachterverblindeten, aktiv kontrollierten Phase-III-Studie an 8015 Personen ab 50 Jahren, die die Immunogenität und
Sicherheit des neuen Kombinationsimpfstoffs untersuchte [5]. Alle Personen erhielten 1:1 randomisiert entweder eine intramuskuläre Injektion mit mCOMBRIAX (+ gleichzeitige Injektion mit
Placebo) oder eine Injektion mit dem COVID-19-Impfstoff Spikevax (mRNA-1273), gleichzeitig verabreicht mit einem zugelassenen quadrivalenten saisonalen Grippeimpfstoff. Personen im Alter von 50-64
Jahren wurden mit FLUARIX (Standarddosis) geimpft, in der Altersgruppe ≥ 65 Jahre wurde hingegen Efluelda (Hochdosis) eingesetzt. Zur Beurteilung der Immunogenität wurden 28 Tage nach der Impfung die
Antikörpertiter mittels Hämagglutinationshemmtest bzw. Pseudovirus-Neutralisationstest sowie die Serokonversion von Anti-Hämagglutinin-Antikörpern und Seroresponse von neutralisierenden Antikörpern
bestimmt. Für mCOMBRIAX konnte gegenüber allen enthaltenen Virusstämmen eine Nichtunterlegenheit der durch die Impfung ausgelösten Immunantworten im Vergleich zur Applikation der jeweiligen
Einzelimpfstoffe gezeigt werden. Darüber hinaus war die durch mCOMBRIAX ausgelöste Immunantwort gegen SARS-CoV-2 (alle Altersgruppen) und alle vier (bei 50-64-Jährigen) bzw. drei (bei ≥ 65-Jährigen)
Influenzastämme stärker als nach der einzelnen Applikation der Impfstoffe. Der einzige Stamm, für den bei Erwachsenen ab 65 Jahren keine statistisch signifikant höhere Immunantwort beobachtet werden
konnte, war B/Yamagata, der entsprechend aktuellen Empfehlungen nicht mehr in saisonalen Grippeimpfstoffen enthalten ist.
Die Impfung mit mCOMBRIAX zeigte ein akzeptables Sicherheits- und Verträglichkeitsprofil. Im Vergleich zur Applikation der Einzelimpfstoffe waren Häufigkeit und Schwere der berichteten Nebenwirkungen
in beiden Alterskohorten zwar numerisch höher (≥ 65 Jahre: 83,5 % und 78,1 %; 50–64 Jahre: 85,2 % und 81,8 %), die meisten waren jedoch von Schweregrad 1 und 2 und von kurzer Dauer [5]. Die
häufigsten Nebenwirkungen von mCOMBRIAX sind Schmerzen an der Injektionsstelle, Müdigkeit, Muskelschmerzen, Gelenkschmerzen, Kopfschmerzen, Schüttelfrost, geschwollene Lymphknoten, Übelkeit und
Erbrechen sowie Fieber [3].
Als erster kombinierter COVID-19/Influenza-Impfstoff bietet mCOMBRIAX Personen ab 50 Jahren die Möglichkeit, sich mit einer einzigen jährlichen Impfung gegen beide Krankheiten zu schützen, wofür
derzeit zwei Injektionen mit Einzelimpfstoffen nötig sind. Für die Zulassungserteilung in Europa ist noch die Zustimmung der Europäischen Kommission ausstehend. Die zum Zeitpunkt der klinischen
Studien untersuchte Impfstoffzusammensetzung (Omicron XBB.1.5, A/H1N1, A/H3N2, B/Victoria, B/Yamagata) basierte auf den Empfehlungen der Weltgesundheitsorganisation für die Saison 2023/2024. Analog
anderen COVID-19- und Grippeimpfstoffen wird auch bei mCOMBRIAX eine jährliche Anpassung an die zirkulierenden Virusstämme erwartet [3].
1 Moderna, Inc. Moderna Receives European Commission Marketing Authorization for COVID-19 Vaccine mNEXSPIKE. Published online Feb 17, 2026. URL: https://feeds.issuerdirect.com/news-release.html?newsid=8201845178897387&symbol=MRNA
2 European Medicines Agency (EMA). mCOMBRIAX – summary of opinion. EMADOC-1829012207-43023. Published online Feb 26, 2026. URL: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-mcombriax_en.pdf
3 European Medicines Agency (EMA). First combined COVID-19 and influenza vaccine for people 50 years and older. Published online Feb 27, 2026. URL: https://www.ema.europa.eu/en/news/first-combined-covid-19-influenza-vaccine-people-50-years-older
4 Robert Koch-Institut (RKI). ARE-Wochenbericht KW 9/2026. doi: 10.25646/13921. URL: https://edoc.rki.de/bitstream/handle/176904/13486/ARE-Wochenbericht_KW09_2026.pdf?sequence=1&isAllowed=y
5 Rudman Spergel, A.K. et al. Immunogenicity and Safety of Influenza and COVID-19 Multicomponent Vaccine in Adults ≥50 Years: A Randomized Clinical Trial. JAMA 2025; 333(22):1977-1987. doi: 10.1001/jama.2025.5646
11.03.2026
Antibiotikatherapie schwerer Infektionen mit multiresistenten Bakterien – Neue S3-Leitlinie
Die Deutsche Gesellschaft für Hygiene und Mikrobiologie (DGHM) hat gemeinsam der Paul-Ehrlich-Gesellschaft für Infektionstherapie, der Deutschen Gesellschaft für Infektiologie und der Österreichischen Gesellschaft für Infektiologie und Tropenmedizin sowie weiteren Fachgesellschaften eine neue S3-Leitlinie zur Antibiotikatherapie schwerer Infektionen mit multiresistenten Bakterien entwickelt. Die Leitlinie gibt evidenzbasierte Empfehlungen für die Behandlung dieser zunehmend herausfordernden Infektionen.
Diagnostik als Grundlage
Ein zentraler Aspekt der Leitlinie ist die Betonung einer qualifizierten mikrobiologischen Diagnostik. Bei der erstmaligen Charakterisierung eines Isolats mit Verdacht auf Multiresistenz während
eines stationären Aufenthaltes soll der Einsatz von jeweils zwei Verfahren zur Erregeridentifizierung und zur Bestimmung der den MRE-Status bestimmenden Resistenzen erfolgen (Empfehlung 2.1.1). Bei
schweren MRE-Infektionen sollten Labororganisation und Verfahren so gestaltet werden, dass die Zeitdauer von der Gewinnung des Untersuchungsmaterials bis zur vorläufigen Befundmitteilung möglichst
kurzgehalten wird (Empfehlung 2.1.2). Bei Verdacht auf eine schwere MRE-Infektion kann eine mikrobiologische Schnelldiagnostik direkt aus dem Untersuchungsmaterial wie Blut oder Liquor mit
Nukleinsäure-basierten Testmethoden erwogen werden (Empfehlung 2.1.3).
Von besonderer Bedeutung bei multiresistenten Gram-negativen Bakterien ist die Produktion von Carbapenemasen. Bei schweren MRE-Infektionen durch Gram-negative Bakterien soll eine Bestimmung des
Resistenzmechanismus, mindestens aber ein Nachweis oder Ausschluss von Metallo-β-Lactamasen (MBL) und gegebenenfalls der nachgewiesenen Carbapenemasen, erfolgen (Empfehlung 2.1.4).
Therapeutische Empfehlungen
Die Leitlinie gibt differenzierte Empfehlungen für die wichtigsten multiresistenten Erreger:
| Erreger | Erste Wahl | Alternative/Besonderheiten |
| CRE (Carbapenem-resistente Enterobacterales) | Ceftazidim-Avibactam, Meropenem-Vaborbactam oder Imipenem-Relebactam (sofern keine MBL) |
Bei Resistenz oder MBL-Nachweis: Cefiderocol oder Aztreonam-Avibactam. |
| DTR-P. aeruginosa (Difficult-to-Treat Resistance) |
Ceftolozan-Tazobactam (bevorzugt unter ABS-Aspekten) |
Ceftazidim-Avibactam, Imipenem-Relebactam, Cefiderocol (bei MBL vorbehalten).
|
| CRAB (Carbapenem-resistente A. baumannii) | Cefiderocol | Sulbactam-Durlobactam in Kombination mit Carbapenem (keine EU-Zulassung). Alternativen: Sulbactam kombiniert mit Levofloxacin, Tigecyclin oder Colistin |
| MRSA-Blutstrominfektion | Daptomycin oder Vancomycin i.v. (Auswahl nach Infektionsfokus und Nephrotoxizitätsrisiko) | Bei Kontraindikation: Teicoplanin, Linezolid oder Ceftobiprol |
| MRSA-Endokarditis | Daptomycin oder Vancomycin i.v. | Bei Kunstklappen: zusätzlich Fosfomycin oder Rifampicin |
| MRSA-Pneumonie | Linezolid oder Vancomycin i.v. | Auswahl nach Toxizitätsrisiko und Co-Medikation |
| MRSA-Haut-/Weichgewebe | Linezolid oder Daptomycin | Tedizolid, Ceftarolin, Ceftobiprol, Delafloxacin oder Vancomycin/Teicoplanin plus Rifampicin/Fosfomycin. Sequenz: Dalbavancin oder Oritavancin |
| VRE-BSI (Vancomycin-resistente Enterokokken) | Linezolid i.v. oder Daptomycin (10 bis 12 mg/kg/Tag) |
Daptomycin nicht bei MHK > 2 mg/l. Tigecyclin/Eravacyclin nicht bei BSI. |
Die Definition der DTR bei P. aeruginosa wurde 2021 von Tamma et al. eingeführt und beschreibt eine gleichzeitige Nichtempfindlichkeit gegenüber allen klinisch relevanten antipseudomonalen Erstlinienantibiotika. Im Unterschied zu den früheren Klassifikationen MDR, XDR und PDR bildet die DTR-Kategorie therapieentscheidende Resistenzmuster praxisnäher ab.
Zur evidenzbasierten Weiterentwicklung der Therapie von Infektionen durch multiresistente Erreger sind prospektive, randomisierte Studien mit klar definierten mikrobiologischen Einschlusskriterien
dringend erforderlich. Besonderes Augenmerk sollte auf die vergleichende Wirksamkeit neuer Substanzen, den Stellenwert von Kombinationstherapien, die optimale Dosierung mit therapeutischem Drug
Monitoring sowie den Einsatz molekularer Diagnostik gelegt werden. Da Erreger und ihre Resistenzen ständigen Veränderungen unterliegen, kann die Leitlinie nur den Stand bis zu ihrem Erscheinen
abbilden.
11.03.2026
Welttuberkulosetag 2026: Ja! Wir können TB beenden!
Die WHO hat den Welttuberkulosetag 2026 unter das Motto „Yes! We can End TB! Led by countries. Powered by people.“ (zu deutsch: „Ja! Wir können TB besiegen! Angeführt von Ländern. Angetrieben von Menschen.“) gestellt. Der am 24. März begangene Aktionstag unterstreicht die zentrale Bedeutung nationaler Strategien und zivilgesellschaftlichen Engagements im Kampf gegen Tuberkulose.
Tuberkulose bleibt eine der führenden infektiösen Todesursachen weltweit. Mit der End-TB-Strategie verfolgt die WHO das Ziel, die Tuberkulose-Epidemie bis 2030 zu beenden – dies bedeutet eine Reduktion der Tuberkulose-bedingten Sterblichkeit um 90 % und der Inzidenz um 80 % im Vergleich zu 2015.
Die Kampagne betont die essenzielle Rolle der einzelnen Länder bei der Implementierung evidenzbasierter Tuberkuloseprogramme sowie die unverzichtbare Bedeutung betroffener Menschen und Gemeinschaften
als treibende Kräfte für Veränderung. Besondere Aufmerksamkeit gilt vulnerablen Populationen mit erhöhtem Tuberkuloserisiko, einschließlich Menschen mit HIV-Koinfektion, Diabetes mellitus und
Unterernährung.
Zur Unterstützung stellt die WHO verschiedene Ressourcen bereit: Der Global TB Report 2025 liefert umfassende epidemiologische Daten, die WHO Academy bietet spezialisierte Kurse für medizinisches
Personal, und am 18. März findet eine Online-Talkshow statt. Die Entwicklung neuer diagnostischer Verfahren, therapeutischer Optionen und die Bekämpfung multiresistenter Tuberkulose bleiben
wissenschaftliche Prioritäten.
>> Mehr Informationen zum Report 2025, zu den Kursen und zur Online-Talkshow
11.03.2026
Leitlinien zur präoperativen Dekolonisierung und Prophylaxe bei multiresistenten Gram-positiven Erregern
Die Europäische Gesellschaft für Klinische Mikrobiologie und Infektionskrankheiten (ESCMID) hat Leitlinien zur präoperativen Dekolonisierung und gezielten Antibiotikaprophylaxe bei erwachsenen Patienten mit multiresistenten Gram-positiven Bakterien (MDR-GPB) vor Operationen veröffentlicht. Die Empfehlungen basieren auf einer systematischen Literaturübersicht und wurden nach dem GRADE-System bewertet.
Für MRSA-Träger ergaben sich folgende Hauptempfehlungen:
- Ein Screening auf S. aureus vor elektiven Herz- und orthopädischen Operationen wird als gute klinische Praxis empfohlen (unbewertet).
- Eine Dekolonisierung mit Mupirocin ± Chlorhexidin wird vor Herz- und orthopädischen Operationen stark empfohlen (moderate Evidenz), für andere Operationen bedingt empfohlen (niedrige Evidenz).
- Eine gezielte perioperative Prophylaxe wird vor Herz-, orthopädischen und neurochirurgischen Eingriffen bedingt empfohlen (niedrige Evidenz).
- Kombinierte Interventionen (Dekolonisierung plus gezielte Prophylaxe) werden vor Herz- und orthopädischen Operationen bedingt empfohlen (sehr niedrige Evidenz).
Für VRE, Methicillin-resistente koagulase-negative Staphylokokken und pan-resistente Gram-positive Bakterien konnten aufgrund unzureichender Evidenz keine Empfehlungen gegeben werden.
| Intervention | Art der OP | Empfehlungsstärke | Evidenzgrad |
| Screening auf S. aureus | Herz-/orthopädische OP | Gute klinische Praxis | unbewertet |
| Dekolonisierung mit Mupirocin ± Chlorhexidin | Herz-/orthopädische OP | Stark | moderat |
| Dekolonisierung mit Mupirocin ± Chlorhexidin | andere OPs | Bedingt | niedrig |
| Gezielte Prophylaxe | Herz-/orthopädische/Neuro-OP | Bedingt | niedrig |
| Kombinierte Interventionen | Herz-/orthopädische OP | Bedingt | sehr niedrig |
Die Autoren betonen, dass die Umsetzung der Empfehlungen an die lokale Epidemiologie und verfügbare Ressourcen angepasst werden sollte. Wichtige Forschungslücken bestehen insbesondere bei:
- Entwicklung von Mupirocin-Resistenzen
- Alternativen Dekolonisierungsstrategien
- Optimaler Teicoplanin-Dosierung
- Interventionen zur VRE-Prävention bei Hochrisikopatienten
Die Leitlinien unterstreichen die Bedeutung eines multidisziplinären Ansatzes zur Infektionsprävention, der neben spezifischen Interventionen bei MDR-GPB-Trägern auch allgemeine Maßnahmen wie
Personalschulung, optimale OP-Techniken und Antibiotic Stewardship umfasst.
Folgerung der Autoren
Die Reduktion postoperativer Infektionen erfordert einen umfassenden Ansatz mit Schulung aller Beteiligten, bewährten chirurgischen Praktiken und einem sorgfältigen Antibiotikamanagement. Die Empfehlungen müssen im Kontext lokaler Gegebenheiten und unter Berücksichtigung möglicher Resistenzentwicklungen umgesetzt werden.
11.02.2026
ESCMID/EUCIC-Leitlinien zur perioperativen Antibiotikaprophylaxe bei MDR-GNB-kolonisierten Patienten
Die Europäische Gesellschaft für Klinische Mikrobiologie und Infektionskrankheiten (ESCMID) und das Europäische Komitee für Infektionsprävention und -kontrolle haben Leitlinien zur perioperativen Antibiotikaprophylaxe (PAP) bei Patienten veröffentlicht, die vor einer Operation mit multiresistenten Gram-negativen Bakterien (MDR-GNB) kolonisiert sind. Die evidenzbasierten Empfehlungen basieren auf einer systematischen Überprüfung von Studien von Januar 2010 bis April 2022.
Die Leitlinien adressieren das Screening auf MDR-GNB vor Operationen, die Anpassung der PAP bei kolonisierten Patienten und die optimale Dauer der PAP. Die Qualität der Evidenz und die Stärke der Empfehlungen wurden nach dem GRADE-Ansatz bewertet.
Wichtige Empfehlungen:
| PAP-Empfehlung MDR-GNB | Screening-Empfehlung | PAP-Empfehlung |
| ESCR-E | Rektales Screening vor kolorektalen OPs und Lebertransplantationen (bedingt, niedrige Evidenz) | Gezielte PAP empfohlen (bedingt, niedrige bis sehr niedrige Evidenz) |
| CRE | Rektales Screening vor Lebertransplantationen (bedingt, niedrige Evidenz) | Keine Empfehlung aufgrund unzureichender Evidenz |
| CRAB | Rektales Screening vor Lebertransplantationen (bedingt, niedrige Evidenz) | Keine Empfehlung aufgrund unzureichender Evidenz |
| FQR-E | Rektales Screening vor TRUSPB (bedingt, moderate Evidenz) | Gezielte PAP empfohlen (bedingt, moderate Evidenz) |
Allgemeine Empfehlungen:
- Screening innerhalb von 3 Wochen vor der Operation (Good Practice)
- PAP-Beendigung innerhalb von 24 Stunden nach OP (stark, moderate Evidenz)
- Bei Transplantationen (außer Niere) PAP-Verlängerung auf 48-72 Stunden möglich (Good Practice)
Die Leitlinien betonen die Notwendigkeit einer sorgfältigen Abwägung zwischen Unter- und Überdiagnose von MDR-GNB-Kolonisationen. Die Implementierung von Screening-Verfahren sollte die lokale
Epidemiologie, mikrobiologische Kapazitäten und verfügbare Ressourcen berücksichtigen.
Folgerung der Autoren
Erhebliche Wissenslücken wurden identifiziert. Gut durchgeführte randomisierte kontrollierte Studien sind dringend erforderlich, insbesondere für CRE- und CRAB-Träger bei Hochrisiko-Operationen.
Zukünftige Studien sollten die Wirksamkeit gezielter PAP und die postoperative MDR-GNB-Kolonisation untersuchen. Ein umfassender Ansatz zur Reduzierung von Wundinfektionen, der sowohl
Antibiotika-basierte Interventionen als auch bewährte chirurgische Praktiken einschließt, wird empfohlen.
11.02.2026
Kommentar der Herausgeber zu beiden Beiträgen
Diese Leitlinien wurden lange erwartet. Die zunehmende Resistenzentwicklung und die wachsende Zahl von Patienten mit multiresistenten Erregern machen deutlich, dass pauschale perioperative
Antibiotikastrategien an ihre Grenzen gestoßen sind. Die neuen Empfehlungen der European Society of Clinical Microbiology and Infectious Diseases (ESCMID) schließen hier eine wichtige Lücke, indem
sie das Problem gezielt aus der Perspektive multiresistenter Erreger adressieren.
Auch in Deutschland zeichnet sich ein Paradigmenwechsel ab: Die aktuelle S3-Leitlinie der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften empfiehlt zunehmend eine individualisierte perioperative Antibiotikaprophylaxe. Die ESCMID-Leitlinien gehen jedoch einen entscheidenden Schritt weiter, indem sie differenziert nach MDR-GNB- und MDR-GPB-Kolonisationen, Operationsart und Evidenzlage vorgehen und damit die klinische Realität in Hochrisikokollektiven besser abbilden.
Gleichzeitig legen die Leitlinien schonungslos die bestehenden Defizite offen. Insbesondere fehlt weiterhin ein tragfähiges Konzept zur Sanierung der intestinalen Kolonisation mit VRE und
multiresistenten gramnegativen Erregern. Für diese Patienten bleibt die perioperative Strategie ein Balanceakt zwischen Infektionsprävention und Vermeidung weiterer Resistenzselektion.
Unser Fazit: Die ESCMID-Leitlinien setzen einen überfälligen, wichtigen Akzent. Sie stärken die individualisierte Entscheidungsfindung, ohne falsche Sicherheit zu suggerieren – und unterstreichen
zugleich den dringenden Bedarf an gezielter klinischer Forschung, gerade dort, wo wir bislang keine wirksamen Sanierungsstrategien haben.
11.02.2026
INFEKTIO_Podcast | Neues aus den Leitlinien – Globale Leitlinie zu Diagnose und Management der Candidiasis
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuelle Folge ist Prof. Oliver Cornely aus Köln zu Gast. Gemeinsam widmen sie sich der globalen Leitlinie zur Diagnose und Management der Candidiasis.
11.02.2026
GeoSentinel-Analyse der Antibiotikaresistenzmuster bei Reisedurchfall
Reisedurchfall ist eine häufige Erkrankung bei internationalen Reisenden, die je nach Reiseziel, Dauer und Jahreszeit 20 % bis 88 % der Reisenden betrifft. In einer aktuellen Querschnittsstudie des GeoSentinel-Netzwerks wurden die Antibiotikaresistenzmuster von vier häufigen Durchfallerregern bei 859 internationalen Reisenden mit akutem Durchfall untersucht, die zwischen April 2015 und Dezember 2022 an 58 Standorten erfasst wurden. Das mediane Alter der Patienten betrug 30 Jahre, 51 % waren männlich. Die häufigsten Reiseregionen waren Subsahara-Afrika (25 %), Südostasien (24 %) und Südamerika (17 %).
Die Analyse der Resistenzmuster zeigte folgende Ergebnisse:
- Campylobacter-Isolate
- Hohe Fluorchinolonen-Resistenz: 75 %
- Höchste Resistenzraten bei Reisenden nach Südzentralasien (88 %) und Südostasien (80 %)
- Makrolid-Resistenz: 12 %
- Höchste Makrolid-Resistenzraten in Südzentralasien (24 %) und Südostasien (11 %)
- Non-Typhus Salmonellen (NTS)-Isolate
- Fluorchinolonen-Resistenz: 32 %
- Höchste Fluorchinolon-Resistenzraten in der Karibik (62 %) und Südzentralasien (45 %)
- Makroliden-Resistenz: 16 %
- Resistenz gegen Cephalosporine der dritten Generation: 5 %
- Shigella-Isolate
- Fluorchinolonen-Resistenz: 22 %
- Auffällig hohe Fluorchinolon-Resistenz von 79 % bei Fällen aus Südzentralasien
- Makroliden-Resistenz: 35 %
- Resistenz gegenüber Cephalosporinen der dritten Generation: 8 %
- Hohe Makrolid-Resistenz von 78 % bei Fällen aus Südamerika
- Diarrhöische E. coli-Stämme
- Fluorchinolon-Resistenz: 18 %
- Resistenz gegenüber Cephalosporinen der dritten Generation: 13 %
- Die meisten E. coli-Isolate stammten von einem einzelnen Standort in Peru
Die Studie zeigt deutliche regionale Unterschiede in den Resistenzmustern. Aufgrund der hohen Fluorchinolon-Resistenz bei Campylobacter und Shigella in Süd- und Südostasien erscheint die Empfehlung
von Makroliden als First-Line-Therapie für diese Regionen gerechtfertigt. In Südamerika, wo eine hohe Makrolid-Resistenz bei Shigella auftrat, kann eine breitere antibiotische Abdeckung erforderlich
sein. In Subsahara-Afrika, wo Campylobacter-Infektionen häufig waren, sind Makrolide aufgrund der 60%igen Fluorchinolon-Resistenz ebenfalls zu bevorzugen.
Folgerung der Autoren
Die Ergebnisse unterstreichen die Bedeutung einer kontinuierlichen Überwachung der Antibiotikaresistenz bei Durchfallerregern durch kulturelle Anzucht und Resistenztestung. Dies ist besonders wichtig angesichts der zunehmenden Verwendung kulturunabhängiger Diagnostikmethoden, die keine Resistenzdaten liefern. Eine sorgfältige Antibiotika-Stewardship und eine gezielte empirische Therapie basierend auf regionalen Resistenzmustern sind entscheidend, um die Entwicklung und Ausbreitung von Antibiotikaresistenzen einzudämmen.
Kommentar der Herausgeber
Die Daten belegen, dass bei Reiserückkehrern aus entsprechenden Risikoregionen (insbesondere Südostasien aber teils auch Lateinamerika) bei Diarrhö oftmals resistente Erreger zugrunde liegen können und eine antibiotische Therapie schwierig ist. Wichtig: In der Regel sind Antibiotika bei Reisediarrhö nicht erforderlich – meist reicht eine orale Rehydrierung aus. Eine Reisediarrhö sollte nur behandelt werden, wenn sie schwer ist, bei blutigen Stühlen, Anzeichen einer systemischen Infektion (Fieber) oder bei immunsupprimierten Patienten.
Amatya, B. et al. GeoSentinel Analysis of Traverls‘ Diarrhea Antimicrobial Restistance Patterns. JAMA Netw Open. 2025; 8(12):e2551089. doi: 10.1001/jamanetworkopen.2025.51089
14.01.2026
Der „One Health“-Zoonose-Bericht 2024 der Europäischen Union
Die Europäische Behörde für Lebensmittelsicherheit (EFSA) und das Europäische Zentrum für die Prävention und die Kontrolle von Krankheiten (ECDC) haben ihren jährlichen „One Health“-Zoonose-Bericht 2024 veröffentlicht. Er fasst die Ergebnisse der Überwachungsaktivitäten zu Zoonosen und zoonotischen Erregern bei Menschen, Tieren, Lebensmitteln und Futtermitteln zusammen, die in den EU-Mitgliedstaaten, dem Vereinigten Königreich (Nordirland) sowie weiteren Ländern erhoben wurden.
Der Bericht beschreibt mehrere zentrale Zoonosen, die in der folgenden Tabelle in abnehmender Häufigkeit dargestellt sind und sich hinsichtlich Krankheitslast und klinischer Bedeutung deutlich unterscheiden.
| Zoonose | Bestätigte Fälle | Melderate pro 100.000 Einwohner | Besonderheiten |
| Campylobacteriose
|
168.396 | 55,3 | Häufigste gemeldete Zoonose; Hauptquelle Geflügelfleisch |
| Salmonellose | 79.703 | 18,6 | Zweithäufigste Zoonose; häufige Ursache lebensmittelbedingter Ausbrüche, v. a. Eier |
| Shiga-Toxin-produzierende Escherichia coli (STEC)-Infektionen | 11.738 | 3,5 | Relevante Ursache schwerer gastrointestinaler Erkrankungen |
| Listeriose Schwerwiegendste Zoonose mit höchstem Anteil an Krankenhausaufenthalten (97,3 %) und 301 Todesfällen | 3.041 | 0,69 | Schwerwiegendste Zoonose; höchste Hospitalisierungs- (97,3 %) und Sterblichkeitsrate (15,6 %) |
Für alle vier Zoonosen wurde in den vergangenen fünf Jahren ein signifikanter Anstieg der gemeldeten Fallzahlen beobachtet.
Auch die Zahl lebensmittelbedingter Ausbrüche nahm 2024 weiter zu: Im Jahr 2024 wurden insgesamt 6.558 Ausbrüche gemeldet, die zu 62.481 Krankheitsfällen, 3.336 Krankenhausaufenthalten und 53 Todesfällen führten. Salmonella war der am häufigsten identifizierte Erreger bei Ausbrüchen mit bekanntem Verursacher (1.238 Ausbrüche) und verursachte die meisten Krankenhauseinweisungen (1.823) sowie zusammen mit Listeria monocytogenes die meisten Todesfälle (jeweils 17). Noroviren und andere Caliciviren waren für die höchste Zahl an Krankheitsfällen verantwortlich (14.297). Die Kombination von Salmonella in „Eiern und Eiprodukten“ wurde als besonders problematisch identifiziert und verursachte die meisten Ausbrüche mit starker Evidenz (83 Ausbrüche).
Im Bereich der Tiergesundheit erfüllten 14 Mitgliedstaaten sowie das Vereinigte Königreich die Zielvorgaben zur Reduzierung der Salmonella-Prävalenz in Geflügelpopulationen. Mehrere Staaten verfehlten jedoch weiterhin die Vorgaben in einzelnen Geflügelkategorien. Auffällig waren zudem deutliche Unterschiede zwischen amtlichen Kontrollen und Eigenkontrollen der Lebensmittelunternehmer: Behördlich entnommene Proben wiesen bei Campylobacter und Salmonella deutlich höhere Überschreitungs- bzw. Prävalenzraten auf als Eigenkontrollen.
Der Bericht enthält zudem aktuelle Daten zu Brucellose, Echinokokkose, Trichinellose, Tuberkulose durch Mycobacterium bovis und Tollwut sowie zu anderen zoonotischen Bakterien, Viren und Parasiten in Lebensmitteln und bei Tieren.
>> Hier geht es zum gesamten Bericht
14.01.2026
INFEKTIO_Podcast | Ersatzteilprobleme – Fremdkörperassoziierten Infektionen
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuelle Folge ist Prof. Andrej Trampuz aus Brisbane (Australien) zu Gast. Gemeinsam widmen sie sich dem Thema fremdkörperassoziierte Infektionen.
14.01.2026
β-Lactam-Allergie-Label und ihre klinischen Auswirkungen
β-Lactam-Antibiotika gehören zu den am häufigsten eingesetzten Antibiotikaklassen, allerdings werden bei 10–15 % der erwachsenen Bevölkerung weltweit β-Lactam-Allergie-Label (BAL) dokumentiert. Diese Label sind jedoch überwiegend nicht verifiziert – bei mehr als 95 % handelt es sich nicht um echte IgE-vermittelte Allergien mit Anaphylaxie und über 90 % der als allergisch gekennzeichneten Patienten vertragen β-Lactame.
Eine aktuelle systematische Übersicht und Meta-Analyse untersuchte die Assoziation zwischen BAL und klinischen Outcomes. Die Analyse basierte auf einer systematischen Literaturrecherche in PubMed, der Cochrane Central Register of Controlled Trials (CENTRAL) und Embase für den Zeitraum Januar 2000 bis November 2024. Eingeschlossen wurden 63 Beobachtungs- und Interventionsstudien, die klinische Outcomes bei Patienten mit und ohne BAL verglichen.
Von den eingeschlossenen Studien stammten 60 (95 %) aus Ländern mit hohem Einkommen, 41 (65 %) aus Amerika, 15 (24 %) aus Europa und 7 (11 %) aus der westpazifischen Region. Die Stichprobengrößen variierten zwischen 86 und 36.856.032 Patienten. 52 Studien (82 %) wurden im stationären Setting durchgeführt und 59 (94 %) untersuchten erwachsene Patienten.
Die Meta-Analyse zeigte signifikante Assoziationen zwischen BAL und verschiedenen negativen klinischen Outcomes:
- Ein um 60 % erhöhtes Risiko für postoperative Wundinfektionen (Odds Ratio [OR] 1,60; 95-%-Konfidenzintervall [KI] 1,27–2,01)
- Ein um 42 % erhöhtes Risiko für Infektionen oder Kolonisation mit multiresistenten Erregern (OR 1,42; 95-%-KI 1,22–1,64)
- Ein um 26 % erhöhtes Risiko für Clostridioides-difficile-Infektionen (OR 1,26; 95-%-KI 1,16–1,37)
Während kein signifikanter Zusammenhang mit der Gesamtmortalität, der Krankenhaussterblichkeit oder der 30-Tage-Sterblichkeit gefunden wurde, zeigte sich bei der Mortalität nach ≥ 180 Tagen ein um
38 % erhöhtes Risiko (OR 1,38; 95-%-KI 1,04–1,85). Die Krankenhausverweildauer war bei Patienten mit BAL statistisch signifikant länger (standardisierte mittlere Differenz 0,06 Tage; 95-%-KI
0,05–0,08) bei jedoch fraglich klinischer Relevanz.
Folgerung der Autoren
Die vorliegende Meta-Analyse zeigt, dass BAL mit einer Reihe von negativen Gesundheitsoutcomes assoziiert sind, insbesondere mit postoperativen Wundinfektionen sowie Infektionen oder Kolonisation
mit multiresistenten Erregern und C. difficile. Obwohl BAL auch statistisch mit längeren Krankenhausaufenthalten assoziiert waren, war der beobachtete Unterschied minimal. Die Heterogenität
und methodischen Limitationen der eingeschlossenen Studien könnten die Generalisierbarkeit und Robustheit einiger Schlussfolgerungen einschränken. Die Ergebnisse unterstreichen jedoch die
Notwendigkeit, Public-Health-Initiativen zur Reduktion inakkurater Allergie-Label zu entwickeln und zu evaluieren, um die unnötige Vermeidung von First-Line-β-Lactam-Antibiotika zu reduzieren.
Kommentar der Herausgeber
Einmal mehr unterstreicht eine große Studie, wie wichtig es in der klinischen Praxis ist, bei allen Patienten eine vermeintliche Antibiotikaallergie zu überprüfen. Wenngleich die vorliegende Arbeit
nur Assoziationen und keine Kausalität etablieren konnte, so sind dennoch die Resultate plausibel und die verschiedenen negativen Auswirkungen erheblich. Hier besteht noch ein enormes
Verbesserungspotential.
Fu, M. et al. The burden of β-lactam allergy labels in health care: a systematic review and meta-analysis. Lancet Infect Dis. 2025; 25(8):896-908. doi: 10.1016/S1473-3099(25)00019-2
17.12.2025
Zulassungsempfehlung für monovalenten Pertussis-Impfstoff
Der Ausschuss für Humanarzneimittel (CHMP) bei der europäischen Arzneimittelbehörde EMA gab nach seiner Sitzung am 13. November 2025 eine positive Stellungnahme für die Zulassung von VacPertagen ab, einem neuen Impfstoff zum Schutz vor Pertussis (Keuchhusten) [1].
Pertussis ist eine hochkontagiöse bakterielle Atemwegserkrankung, die durch Bordetella pertussis verursacht wird. Es besteht keine lebenslange Immunität; sowohl nach natürlicher Infektion als auch nach Impfung nimmt der Schutz innerhalb weniger Jahre deutlich ab, was zu häufigen Reinfektionen im Erwachsenenalter führt [2,3]. In Deutschland liegt das mittlere Alter der gemeldeten Patienten bei etwa 40 Jahren [4,5]. Bei Erwachsenen verläuft die Erkrankung meist mild mit wochenlangem, hartnäckigem Husten, während Säuglinge besonders gefährdet sind und oft einen klassischen Verlauf in drei Stadien zeigen (katarrhalisch, paroxysmal, konvaleszent) [2,6,7]. Die größte Gefahr besteht darin, dass Erwachsene als Hauptreservoir die Infektion auf ungeimpfte Säuglinge übertragen, bei denen schwere Komplikationen und Todesfälle auftreten können [3,7]. Seit März 2013 besteht in Deutschland eine bundesweite Meldepflicht für Pertussis; zuvor war die Meldepflicht auf die östlichen Bundesländer beschränkt [8]. Auch in Österreich ist die Erkrankung meldepflichtig, in der Schweiz hingegen nicht. Im Jahr 2024 wurde dem Robert Koch-Institut die bislang höchste jährliche Fallzahl seit Einführung der Meldepflicht gemeldet, nach einem pandemiebedingten Rückgang in den Vorjahren [9]. Die Infektionsschutzmaßnahmen während der COVID-19-Pandemie führten zu einem deutlichen Rückgang der Pertussis-Fälle, der nach Aufhebung der Maßnahmen wieder anstieg [9].
Zur Prävention werden in Deutschland, Österreich und Schweiz die Impfung von Schwangeren zur passiven Immunisierung des Neugeborenen sowie die Grundimmunisierung im Säuglings- und Kleinkindalter, gefolgt von regelmäßigen Auffrischimpfungen im Kinder-, Jugend- und Erwachsenenalter empfohlen. Die Diagnostik erfolgt bevorzugt mittels PCR [6]. Die konsequente Umsetzung dieser Maßnahmen ist entscheidend, um schwere Verläufe und Todesfälle bei Säuglingen zu verhindern.
Der neue Impfstoff VacPertagen soll als Suspension zur Injektion in einer Fertigspritze erhältlich sein und ist vorgesehen für die Auffrischimpfung gegen Pertussis bei Personen ab 12 Jahren und zum passiven Schutz gegen Pertussis im frühen Säuglingsalter nach mütterlicher Immunisierung während der Schwangerschaft [1]. VacPertagen enthält zwei gereinigte Pertussis-Antigene: rekombinantes Pertussis-Toxin (PTgen) und filamentöses Hämagglutinin (FHA). Nach intramuskulärer Applikation induziert VacPertagen eine Verstärkung der PT- und FHA-spezifischen Antikörperreaktionen. Maternale Antikörper werden auf Säuglinge übertragen, wenn eine Impfung im zweiten oder dritten Schwangerschaftstrimester erfolgt ist.
VacPertagen wurden in drei klinischen Studien untersucht, die zeigten, dass der Impfstoff 28 Tage nach der Impfung bei Erwachsenen und Jugendlichen die Produktion von Antikörpern auslöste, die bei Erwachsenen bis zu drei Jahre und bei Jugendlichen bis zu fünf Jahre anhielten [1]. Diese Immunantwort wurde auch bei schwangeren Frauen 28 Tage nach der Impfung im zweiten oder dritten Trimester ausgelöst. Die bei der Geburt auf die Säuglinge übertragenen Pertussis-Antikörper blieben bis zu einem Alter von zwei Monaten bestehen. Zu den häufigsten Nebenwirkungen von VacPertagen zählen Schmerzen an der Injektionsstelle, Kopfschmerzen, Müdigkeit, Myalgie, Arthralgie, Unwohlsein und Übelkeit.
Folgt die Europäische Kommission der Empfehlung des CHMP und wird VacPertagen in Europa zugelassen, wäre es aktuell der einzige monovalente Pertussis-Impfstoff. Seit der Marktrücknahme früherer monovalenter Impfstoffe in Deutschland vor etwa 20 Jahren stehen für die Immunisierung gegen Pertussis hierzulande, sowie in Österreich und Schweiz, ausschließlich verschiedene Kombinationsimpfstoffe (als 3-, 4-, 5- oder 6-fach-Impfstoffe) zur Verfügung, die gleichzeitig gegen Diphtherie, Tetanus, Poliomyelitis, Hepatitis B und Haemophilus influenzae Typ b schützen.
1 European Medicines Agency (EMA). VacPertagen - summary of opinion. EMA/CHMP/354843/2025; published online Nov 14, 2025. URL: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-vacpertagen_en.pdf
2 Zepp, F. et al. Rationale for Pertussis Booster Vaccination Throughout Life in Europe. Lancet Infect Dis 2011; 11(7):557-70. doi: 10.1016/S1473-3099(11)70007-X
3 Sheng, Y. et al. Pertussis Resurgence: Epidemiological Trends, Pathogenic Mechanisms and Preventive Strategies. Front Immunol. 2025; 16:1618883. doi: 10.3389/fimmu.2025.1618883
4 Hellenbrand, W. et al. The Epidemiology of Pertussis in Germany: Past and Present. BMC Infect Dis. 2009; 9:22. doi: 10.1186/1471-2334-9-22
5 Hitz, D.A. et al. Seasonal Bordetella Pertussis Pattern in the Period From 2008 to 2018 in Germany. BMC Infect Dis. 2020; 20(1):474. doi: 10.1186/s12879-020-05199-w
6 Kline, J.M. et al. Pertussis: Common Questions and Answers. Am Fam Physician 2021; 104(2):186-192. URL: https://www.aafp.org/pubs/afp/issues/2021/0800/p186.html
7 Kandeil, W. et al. A Systematic Review of the Burden of Pertussis Disease in Infants and the Effectiveness of Maternal Immunization Against Pertussis. Expert Rev Vaccines. 2020; 19(7):621-638. doi:
10.1080/14760584.2020.1791092
8 Schielke, A. et al. Marked Underreporting of Pertussis Requiring Hospitalization in Infants as Estimated by Capture-Recapture Methodology, Germany, 2013-2015. Pediatr Infect Dis J. 2018;
37(2):119-125. doi: 10.1097/INF.0000000000001698
9 Gorringe, A. et al. Global Incidence of Pertussis After the COVID-19 Pandemic. JAMA Netw Open. 2025; 8(12):e2545963. doi: 10.1001/jamanetworkopen.2025.45963
17.12.2025
INFEKTIO_Podcast | Vom Bock zum Gärtner – Borrelien richtig diagnostizieren und behandeln
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuellen Folge ist Dr. Volker Fingerle vom nationalen Referenzzentrum für Borrelien zu Gast. Gemeinsam widmen sie sich dem Thema Borrelien-Infektionen.
17.12.2025
Neue Impfempfehlungen für immunsupprimierte Patienten: COVID-19, Influenza und RSV
Die Infectious Diseases Society of America (IDSA) hat neue Leitlinien für die Impfung immunsupprimierter Patienten gegen COVID-19, Influenza und das Respiratorische Synzytialvirus (RSV) veröffentlicht. Die Empfehlungen richten sich an Patienten mit hämatologischen Malignomen, primären Immundefekten, Autoimmunerkrankungen unter Immunsuppressiva oder Biologika sowie HIV-Patienten mit schwerer Immunsuppression. Auch Empfänger von soliden Organtransplantaten, hämatopoetischen Stammzelltransplantationen und CAR-T-Zell-Therapien werden berücksichtigt.
Für die COVID-19-Impfung zeigten Studien eine Impfwirksamkeit von 33 % bis 56 % hinsichtlich der Verhinderung von Krankenhausaufenthalten. Die Wirksamkeit gegen kritische Erkrankungen lag bei 40 % (95-%-Konfidenzintervall [KI] 26–51 %) und gegen COVID-19-bedingte Sterblichkeit bei 61 % (95-%-KI 36–77 %). Schwerwiegende unerwünschte Ereignisse traten selten auf.
Bei der Influenza-Impfung wurde eine Wirksamkeit von 32 % (95-%-KI 7–50 %) gegen influenzabedingte Hospitalisierungen bei immunsupprimierten Erwachsenen nachgewiesen. Die Wirksamkeit gegen Aufnahmen auf Intensivstationen betrug 41 % (95-%-KI 31–50 %). Die Sicherheitsdaten zeigten kein erhöhtes Risiko für das Guillain-Barré-Syndrom oder andere schwerwiegende Nebenwirkungen.
Die RSV-Impfung erwies sich als besonders effektiv mit einer Wirksamkeit von 70 % (95-%-KI 66–73 %) gegen RSV-assoziierte Hospitalisierungen. Gegen kritische Erkrankungen, definiert als Intensivaufnahme oder Tod im Krankenhaus, lag die Wirksamkeit bei 81 % (95-%-KI 52–92 %). Es wurde ein leicht erhöhtes Risiko für das Guillain-Barré-Syndrom mit 11,2 zusätzlichen Fällen pro 1 Million Impfdosen beobachtet.
Die IDSA empfiehlt für alle drei Impfungen eine altersgerechte Verabreichung (starke Empfehlung, moderate Evidenzqualität). Die Impfungen können gleichzeitig verabreicht werden. Für eine optimale Wirksamkeit sollte der Impfzeitpunkt an die individuelle immunsuppressive Therapie angepasst werden. Bei Organtransplantationen wird die Impfung idealerweise mindestens 2 Wochen vor der Transplantation oder frühestens 1–3 Monate danach empfohlen. Nach hämatopoetischer Stammzelltransplantation oder CAR-T-Zell-Therapie sollte mindestens 3 Monate gewartet werden.
Folgerung der Autoren
Die IDSA-Leitlinien unterstreichen die Bedeutung der Impfungen gegen COVID-19, Influenza und RSV bei immunsupprimierten Patienten. Trotz möglicherweise reduzierter Impfantwort überwiegt der Nutzen
deutlich die Risiken. Individuelle Impfstrategien, einschließlich der Berücksichtigung von Therapieplänen und Risikofaktoren, sind essenziell für einen optimalen Schutz dieser vulnerablen
Patientengruppe.
19.11.2025
Kurz gefragt: Wie lesen Sie Fachinformationen?
Fachinformationen lassen sich heute auf ganz unterschiedlichen Wegen nutzen. Daher interessiert uns, wie Sie solche Inhalte in Ihrem Alltag am liebsten lesen – egal ob digital oder gedruckt. Ihre
kurze Rückmeldung hilft uns zu verstehen, über welches Medium Sie Fachinformationen am häufigsten nutzen.
Bitte nehmen Sie sich ca. 1 Minute Zeit, um uns Ihr Feedback mitzuteilen. Als Dank erhalten Sie das Whitepaper zum Thema „Antibiotic Stewardship”.
19.11.2025
ECDC startet neue Berichte und Leitlinien zu mückenübertragenen Krankheiten in Europa
In diesem Jahr hat das Europäische Zentrum für die Prävention und die Kontrolle von Krankheiten (ECDC) eine neue Reihe wöchentlicher Überwachungsberichte gestartet. Diese sollen Gesundheitsbehörden dabei unterstützen, mückenübertragene Krankheiten wie Chikungunya, Dengue, Zika und West-Nil-Virus zeitnah zu überwachen. Durch die Bereitstellung konsistenter, nahezu Echtzeit-epidemiologischer Daten können die Berichte nationale und regionale Kontrollstrategien unterstützen, insbesondere da Europa aufgrund des Klimawandels mit längeren und intensiveren Mückensaisons konfrontiert ist.
Die Hauptvektoren, die in Europa Anlass zur Sorge geben, sind Aedes albopictus, Aedes aegypti und Culex pipiens. Aedes albopictus ist derzeit in 16 Ländern und 369
Regionen etabliert – deutlich mehr als noch vor einem Jahrzehnt. Im vergangenen Jahr wurden in Europa 304 Fälle von lokal erworbenem Dengue-Fieber und 1.436 Fälle von West-Nil-Virus-Infektionen
registriert, was die wachsende Ausbreitung dieser Krankheiten unterstreicht.
Um die Vorbereitungsmaßnahmen weiter zu unterstützen, veröffentlicht das ECDC auch neue Leitlinien für das öffentliche Gesundheitswesen zu lokal erworbenen Aedes-übertragenen Krankheiten in Europa.
Das Dokument beschreibt praktische Überwachungs-, Präventions- und Kontrollmaßnahmen, basierend auf vier Risikoniveaus.
19.11.2025
Weltweite Aktionswoche zur World Antimicrobial Awareness Week 2025
Die Weltgesundheitsorganisation WHO ruft zur World Antimicrobial Resistance Awareness Week 2025 auf, die vom 18. bis zum 24. November stattfindet. Diese globale Kampagne soll das Bewusstsein für antimikrobielle Resistenzen (AMR) schärfen und zu verantwortungsvollem Umgang mit Antibiotika sensibilisieren. Die WHO stellt dafür Kampagnenmaterialien zur Verfügung und lädt zur Teilnahme an der „Go Blue for AMR“-Initiative ein.
19.11.2025
Respiratorische Virusinfektionen aktivieren metastatische Brustkrebszellen in der Lunge
Brustkrebs ist weltweit die häufigste Krebserkrankung bei Frauen, wobei die meisten Todesfälle durch Metastasen verursacht werden. Oft bleiben disseminierte Tumorzellen über lange Zeiträume hinweg in einem Ruhezustand, bevor es zu einer metastatischen Progression kommt. Das Verständnis der Mechanismen, die diesen Ruhezustand durchbrechen, ist entscheidend für die Behandlung von Metastasen. Infektionen durch respiratorische Viren wie Influenza und SARS-CoV-2 lösen sowohl lokale als auch systemische Entzündungsreaktionen aus.
In einer aktuellen Studie konnten Forscher in Mausmodellen zeigen, dass Infektionen mit Influenza- und SARS-CoV-2-Viren zum Verlust des Ruhephänotyps von Brustkrebszellen in der Lunge führen. Dies führte innerhalb weniger Tage nach der Infektion zu einer Proliferation der Krebszellen und innerhalb von zwei Wochen zu einer massiven Expansion zu metastatischen Läsionen. Diese phänotypischen Übergänge und Expansionen erwiesen sich als Interleukin-abhängig.
Die Studie verwendete verschiedene Mausmodelle für Brustkrebs, darunter MMTV-PyMT und MMTV-Her2 Mäuse, die jeweils die Onkogene Polyomavirus mittleres T-Antigen (PyMT) bzw. Her2 exprimieren. MMTV-Her2 Mäuse (FVB) wurden mit IL-6-Knockout (KO) Mäusen gekreuzt. Für ein orthotopes Modell des Brustkrebses wurden E0771 Brustkrebszellen in die vierte rechte und linke Brustdrüse injiziert. Acht Wochen alte MMTV-PyMT und 12-14 Wochen alte MMTV-Her2 weibliche Mäuse wurden mit 500 PFU Influenza A/PR/8/34 H1N1 IAV oder SARS-CoV-2 intranasal infiziert.
Die Studie zeigte, dass virale respiratorische Infektionen (Influenza und SARS-CoV-2) die Aktivierung von Lungen T-Zellen beeinträchtigen und dass CD4+ T-Zellen die pulmonale metastatische Last nach der Influenzainfektion aufrechterhalten, indem sie die Aktivierung und Zytotoxizität von CD8+T-Zellen hemmen. Diese experimentellen Befunde stimmen mit Analysen von Daten zu Krebsüberlebenden aus den US-Datenbanken überein.
Analysen der SEER-Medicare-Datenbank für alle Krebsarten und der Flatiron Health-Datenbank für Brustkrebs zeigten, dass eine SARS-CoV-2-Infektion das Risiko für krebsbedingte Mortalität und Lungenmetastasen im Vergleich zu nicht infizierten Krebsüberlebenden deutlich erhöhte. Bei der Analyse der SEER-Medicare-Daten wurden 13,3 Millionen Medicare-Begünstigte im Alter von 65 Jahren oder älter eingeschlossen, von denen 3,14 Millionen eine Krebsdiagnose hatten. Von diesen hatten 164.507 (5,24 %) eine SARS-CoV-2-Diagnose. In der Flatiron Health-Datenbank wurden 69.256 Brustkrebspatienten analysiert, von denen 5.602 (8,1 %) eine SARS-CoV-2-Diagnose hatten.
Folgerung der Autoren
Diese Entdeckungen unterstreichen den erheblichen Einfluss respiratorischer Virusinfektionen auf das Wiederauftreten metastatischer Krebserkrankungen. Sie liefern neue Erkenntnisse über den Zusammenhang zwischen Infektionskrankheiten und Krebsmetastasen. Die Ergebnisse deuten darauf hin, dass respiratorische Virusinfektionen wie Influenza und SARS-CoV-2 nicht nur eine unmittelbare Gesundheitsbedrohung darstellen, sondern auch langfristige Auswirkungen auf Krebspatienten haben können, insbesondere in Bezug auf die Entwicklung von Lungenmetastasen. Die Autoren betonen die Notwendigkeit weiterer Forschung zur Entwicklung gezielter Präventions- und Behandlungsstrategien für Krebspatienten mit respiratorischen Virusinfektionen.
Kommentar der Herausgeber
Eine weitere spannende Studie, die belegt, dass Infektionen nicht-infektiöse Folgeerkrankungen auslösen oder aggravieren können. Es gibt – neben dem klar belegten Zusammenhang zwischen HPV und Zervixkarzinomen – Daten zu Influenza, RSV und Pneumokokken und kardiovaskulären Ereignissen und Herpes Zoster und Demenz. Solche Daten zu erwünschten Impfwirkungen könnten helfen, die Impfbereitschaft zu steigern.
Chia, S. B. et al. Respiratory viral infections awaken metastatic breast cancer cells in lungs. Nature. 2025; 625(7993):572-581. doi: 10.1038/s41586-024-07003-0
22.10.2025
In eigener Sache
Update der S3-Leitlinie Sepsis 2025: Neue Empfehlungen für die Praxis
Die Deutsche Sepsis-Gesellschaft hat gemeinsam mit 15 weiteren Fachgesellschaften die S3-Leitlinie zur Sepsis grundlegend überarbeitet. Das Update bringt wichtige Neuerungen für Prävention, Diagnose, Therapie und Nachsorge. Besonders bedeutsam sind die Empfehlungen zum frühen Sepsis-Screening, zur antimikrobiellen Therapie und zur Nachsorge von Sepsis-Überlebenden. Die neuen Leitlinien orientieren sich an aktuellen internationalen Standards und bieten konkrete Handlungsempfehlungen für die klinische Praxis.
Lesen Sie hier als Abonnent:in weiter
Sie haben noch kein Abo?
» Hier informieren und Zugang zu allen Fachartikeln erhalten.
Zulassungsempfehlung für weiteren Antikörper zur RSV-Prophylaxe
In Deutschland sind Infektionen mit dem Respiratorischen Synzytial-Virus (RSV) die häufigste Ursache für Krankenhauseinweisungen von Säuglingen, bei denen insbesondere in den ersten sechs Lebensmonaten ein erhöhtes Risiko für schwere Krankheitsverläufe besteht [1]. Seit Juni 2024 empfiehlt die Ständige Impfkommission (STIKO) am Robert Koch-Institut (RKI) daher allen Neugeborenen und Säuglingen in ihrer ersten RSV-Saison eine Prophylaxe mit dem monoklonalen Antikörper Nirsevimab [1]. Dass diese Strategie Wirkung zeigt, legt eine erste Auswertung von Meldedaten aus Deutschland nahe [2]. Seit der Einführung der RSV-Prophylaxe konnte die Krankheitslast bei Säuglingen < 1 Jahr deutlich reduziert werden: im Vergleich zur Vorsaison sank die Inzidenz der an das RKI übermittelten laborbestätigten RSV-Fälle 2024/2025 um 54 %. Nachdem der Ausschuss für Humanarzneimittel (CHMP) bei der europäischen Arzneimittelbehörde EMA im September 2025 das Präparat ENFLONSIA der Firma Merck Sharp Dohme zur Zulassung empfohlen hat, könnte demnächst ein weiterer monoklonaler Antikörper zur RSV-Prophylaxe von Neugeborenen und Säuglingen in Europa zur Verfügung stehen [3].
ENFLONSIA enthält Clesrovimab, ein neutralisierender humaner monoklonaler IgG1κ-Antikörper, der gegen das RSV-Fusionsprotein F gerichtet ist [3]. Das Fusionsprotein auf der Oberfläche des Virus ist maßgeblich an der Infektion des Körpers beteiligt, indem es die Fusion mit den Schleimhautepithelzellen des Respirationstraktes ermöglicht. Die Bindung von Clesrovimab an das Fusionsprotein blockiert den entscheidenden Membranfusionsschritt und verhindert das Eindringen des Virus in die Zellen [3, 4]. Durch eine Modifizierung der Aminosäuresequenz in der FC-Region des Antikörpers ist die Bindung an den neonatalen FC-Rezeptor erhöht und damit die Serumhalbwertszeit auf etwa 44 Tage verlängert [3, 4].
In einer randomisierten, doppelblinden, placebokontrollierten Phase-IIb/III-Studie erhielten 3614 gesunde Säuglinge entweder eine intramuskuläre Einzeldosis von 105mg Clesrovimab (n = 2411) oder Placebo (n = 1203) [5]. Die Inzidenz medizinisch behandelter RSV-Infektionen der unteren Atemwege innerhalb von 150 Tagen nach der Injektion betrug 2,6 % unter Clesrovimab gegenüber 6,5 % unter Placebo, entsprechend einer Wirksamkeit der Prophylaxe von 60,4 % (95 %-Konfidenzintervall (KI) 44,1-71,9 %; p < 0,001). Auch RSV-assoziierte Hospitalisierungen konnten nach der Applikation von Clesrovimab innerhalb des Beobachtungszeitraums signifikant reduziert werden (Wirksamkeit 84,2 %; 95 %-KI 66,6-92,6 %; p < 0,001). Die Sicherheitsprofile waren vergleichbar, wobei schwerwiegende unerwünschte Ereignisse bei 11,5 % der mit Clesrovimab behandelten Säuglinge gegenüber 12,4 % in der Placebogruppe berichtet wurden.
In einer weiteren randomisierten, teilweise verblindeten Phase-III-Studie wurde die Wirksamkeit und Sicherheit einer Einzeldosis Clesrovimab (n = 450) im Vergleich zur monatlichen Applikation von Palivizumab (n = 451) bei Säuglingen und Kindern mit erhöhtem Risiko für schwere RSV-Erkrankungen untersucht [6]. Erste Zwischenergebnisse für den Zeitraum von 150 Tagen nach der (ersten) Injektion zeigten eine ähnliche Inzidenz medizinisch behandelter RSV-Infektionen der unteren Atemwege (3,6 % bzw. 3,0 %) und RSV-assoziierter Hospitalisierungen (1,3 % bzw. 1,5 %) unter Clesrovimab und Palivizumab bei vergleichbarem Sicherheitsprofil.
Zu den häufigsten Nebenwirkungen zählen Schmerzen, Erytheme und Schwellungen an der Injektionsstelle sowie Hautausschlag.
Wenn die Europäische Kommission der positiven Stellungnahme des CHMP folgt und die Zulassung für ENFLONSIA erteilt, ist Clesrovimab der dritte monoklonale Antikörper zur RSV-Prophylaxe in Europa und der erste, der unabhängig vom Gewicht des Säuglings als einmalige Fixdosis verabreicht wird. Clesrovimab bindet an der hochkonservierten Stelle IV des RSV-Fusionsproteins, was sich von den RSV-Antikörpern Palivizumab (Stelle II) und Nirsevimab (Stelle Ø) unterscheidet. Aufgrund der verlängerten Halbwertszeit bietet eine Dosis Clesrovimab, ähnlich wie Nirsevimab, bis zu fünf Monate lang passive Immunität, was die Dauer einer üblichen RSV-Saison in Europa abdeckt.
1 Koch, J. et al. Beschluss und wissenschaftliche Begründung zur Empfehlung der STIKO zur spezifischen Prophylaxe von RSV-Erkrankungen mit Nirsevimab bei Neugeborenen und Säuglingen in ihrer 1. RSV-Saison. Epid Bull 2024; 26:3-29. doi: 10.25646/12198 [24.09.2025]
2 Schönfeld, V. et al. The incidence of RSV infection since the introduction of monoclonal antibody prophylaxis: An analysis of reported case data across Germany for the seasons 2023/24 and 2024/25.
Dtsch Arztebl Int. 2025; 122:472–3. doi: 10.3238/arztebl.m2025.0111
3 European Medicines Agency (EMA). Enflonsia - summary of opinion. EMA/CHMP/274513/2025; published online Sep 19, 2025. URL: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-enflonsia_en.pdf [24.09.2025]
4 U.S. Food & Drug Administration (FDA). ENFLONSIA (Clesrovimab-cfor): Full Prescribing Information. Jun 2025. URL: https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/761432s000lbl.pdf [24.09.2025]
5 Zar, H. J. et al. Clesrovimab for Prevention of RSV Disease in Healthy Infants. N Engl J Med. 2025; published online first Sep 17, 2025. doi: 10.1056/NEJMoa2502984
6 Zar, H. J. et al. Clesrovimab in Infants and Children at Increased Risk for Severe RSV Disease. N Engl J Med 2025; published online first Sep 17, 2025. doi: 10.1056/NEJMc2506107
22.10.2025
Ausbruch von Corynebacterium diphtheriae bei Migranten in Europa
Ein ungewöhnlicher Anstieg von Infektionen mit Corynebacterium diphtheriae wurde ab Sommer 2022 in europäischen Aufnahmezentren für Migranten beobachtet. Ein pan-europäisches Konsortium untersuchte die klinischen, epidemiologischen und mikrobiologischen Merkmale dieses Ausbruchs, der den größten Anstieg von Diphtherie-Fällen in Westeuropa seit 70 Jahren darstellt.
In die Analyse wurden 363 toxigene C. diphtheriae-Isolate von 362 Patienten aus 10 europäischen Ländern einbezogen, die zwischen Januar und November 2022 gemeldet wurden. Die meisten Fälle traten in Deutschland (118), Österreich (66), Großbritannien (59), der Schweiz (52) und Frankreich (30) auf. Das mediane Alter der Patienten betrug 18 Jahre, wobei 176 Patienten (48,6 %) zwischen 16 und 20 Jahre alt waren. Die große Mehrheit der Betroffenen (98,1 %) war männlich. Bei 348 Patienten (96,1 %) lag eine kürzliche Migrationsgeschichte vor oder sie hatten engen Kontakt zu Migrantenpopulationen. 174 Patienten (48,1 %) waren in Migrantenzentren untergebracht.
Klinische Daten lagen für 346 Patienten (95,6 %) vor: 268 Patienten (77,5 %) wiesen eine kutane Diphtherie auf, 53 (15,3 %) eine respiratorische Form (11 davon mit Pseudomembran) und 9 (2,6 %) zeigten sowohl respiratorische als auch kutane Symptome. Ein Patient mit Pseudomembran verstarb. Bei drei Patienten wurde eine genitale Beteiligung festgestellt. Der Impfstatus konnte aufgrund unvollständiger medizinischer Dokumentation häufig nicht zuverlässig ermittelt werden. Nur bei 4 Patienten war eine Impfung dokumentiert, bei 10 Patienten wurde eine fehlende Impfung vermerkt und bei 290 Patienten war der Impfstatus unbekannt.
Von 17 hospitalisierten Patienten hatten 5 eine kutane und 12 eine respiratorische Diphtherie (7 mit Pseudomembran). Alle 12 Patienten mit respiratorischer Diphtherie erhielten Diphtherie-Antitoxin zusammen mit Antibiotika. Die Antitoxin-Dosen lagen zwischen 5000 und 100.000 IE, wobei 8 Patienten Dosen über 60.000 IE erhielten. Die Behandlung erfolgte im Durchschnitt 2,8±1,5 Tage nach Symptombeginn, die meisten (9) Patienten wurden innerhalb von 3 Tagen behandelt.
Die molekulargenetische Analyse identifizierte vier Hauptcluster, was auf die multiklonale Natur des Ausbruchs hinweist. Das ermX-Gen (kodiert für Erythromycin-Resistenz) sowie die Gene pbp2m und blaOXA-2 (kodieren für β-Lactam-Resistenz) wurden in einer Untergruppe von Isolaten nachgewiesen. Isolate mit ermX waren resistent gegen Erythromycin, während Isolate mit pbp2m resistent gegen Penicillin, aber empfindlich gegenüber Amoxicillin waren.
Im Jahr 2023 wurden in den beteiligten Ländern weitere 169 Fälle von C. diphtheriae-Infektionen mit toxigenen Isolaten gemeldet: 112 in Deutschland, 17 in den Niederlanden, 16 in Frankreich, 13 in Großbritannien, 8 in der Schweiz und 3 in Österreich, mit zwei weiteren Todesfällen bei Migranten. Obwohl weniger Fälle als 2022 gemeldet wurden, lag die Gesamtzahl deutlich über den Vorjahren.
Folgerung der Autoren
Die Verteilung der genetischen C. diphtheriae-Cluster über mehrere europäische Länder zeigt eine wiederholte grenzüberschreitende Ausbreitung. Die hohe Anzahl von C. diphtheriae-Infektionen bei Migranten ist besorgniserregend, insbesondere da Antibiotikaresistenz-Phänotypen die Wirksamkeit von First-Line-Behandlungen gefährden. Die Ergebnisse unterstreichen die Notwendigkeit verbesserter Präventionsmaßnahmen, einschließlich gründlicher Impfprotokolle für Migranten und lokale Bevölkerungen, klinische Überwachung von Risikopersonen, schnelle Diagnose bei symptomatischen Personen sowie Screening von Kontaktpersonen.
Kommentar der Herausgeber
Die Diphtherie ist in Deutschland, Österreich und der Schweiz meldepflichtig und kann potenziell tödlich verlaufen. Bei häufig ungeimpften Migranten mit kutanen oder pharyngealen Symptomen ist es wichtig, an diese wichtige Differentialdiagnose zu denken. Entscheidend ist eine rasche Gabe von Antibiotika und, in toxin-positiven Fällen, aber auch bereits bei klinischem Verdacht auf eine respiratorische Diphtherie, frühzeitig von Antitoxin. Bei kutaner Diphtherie wird eine Antitoxin-Gabe in der Regel nicht empfohlen, außer bei Ulcera von mehr als 2 cm². Ebenso essenziell sind die Implementierung infektpräventiver und postexpositioneller Maßnahmen. Dieser multinationale Ausbruch einer alten, beinahe vergessenen Infektion führt eindrücklich vor Augen, wie wichtig eine gute Durchimpfungsquote ist, gerade angesichts grassierender Falschinformationen hinsichtlich der Sicherheit von Impfungen.
Hoefer, A. et al. Corynebacterium diphtheriae Outbreak in Migrant Populations in Europe. N Engl J Med. 2025; 392(23):2334-2345. doi: 10.1056/NEJMoa2311981
10.09.2025
WHO-Bericht deutet auf wahrscheinlichen zoonotischen Ursprung von COVID-19 hin
Ein kürzlich veröffentlichter Bericht der Weltgesundheitsorganisation (WHO) präsentiert neue Erkenntnisse zum Ursprung von COVID-19. Die Wissenschaftliche Beratergruppe für den Ursprung neuartiger Krankheitserreger (SAGO) hat die verfügbaren Beweise ausgewertet und kommt zu dem Schluss, dass ein zoonotischer Ursprung des SARS-CoV-2-Virus am wahrscheinlichsten ist. [1]
Der SAGO-Bericht stützt sich auf begutachtete Studien, Expertenbriefings und unveröffentlichte Daten. Die Gruppe betont, dass ein Übertritt des Virus von Tieren auf den Menschen die plausibelste Erklärung für den Ausbruch darstellt. Allerdings schließt der Bericht die Möglichkeit eines Laborunfalls nicht vollständig aus, da chinesische Behörden Anfragen zu Forschungsaktivitäten und Biosicherheitsverfahren in Laboren in Wuhan nicht beantwortet haben. [1,2]
Als stärkstes Indiz für einen zoonotischen Ursprung wird eine metagenomische Analyse von Tier- und Umweltproben aus dem Huanan-Markt in Wuhan angeführt. Diese bestätigte die Anwesenheit von für
SARS-CoV-2 anfälligen Tierarten in Marktständen mit hoher Virusbelastung. Insbesondere wurden DNA-Spuren von Marderhunden, Bambusratten und Schleichkatzen in Proben mit hoher SARS-CoV-2-Konzentration
nachgewiesen. [2,3]
Der Bericht hebt hervor, dass trotz umfangreicher Forschung weiterhin entscheidende Daten fehlen, um den genauen Zeitpunkt und Ort des ersten Übertritts auf den Menschen zu bestimmen. Es bleibt
unklar, ob der Huanan-Markt tatsächlich der Ursprungsort war oder ob er lediglich als Verstärker für die Virusausbreitung diente. [1,2]
SAGO fordert weitere Untersuchungen, insbesondere zu Tieren und Tierhändlern mit Verbindungen zum Huanan-Markt sowie zu Zuchtbetrieben in der Region. Auch empfiehlt die Gruppe, die Suche nach frühen
COVID-19-Fällen vor Dezember 2019 fortzusetzen. [1,3]
Der Bericht betont die Notwendigkeit internationaler Zusammenarbeit und Transparenz bei der Erforschung von Krankheitsausbrüchen. Er unterstreicht zudem die Bedeutung des „One Health“-Ansatzes, der
die Verbindungen zwischen Mensch, Tier und Umwelt berücksichtigt. [1,2]
Obwohl der zoonotische Ursprung als wahrscheinlichste Hypothese gilt, mahnt SAGO zur Vorsicht bei endgültigen Schlussfolgerungen. Die Gruppe bleibt offen für neue Erkenntnisse und betont die
Wichtigkeit weiterer Forschung zur Klärung des COVID-19-Ursprungs. [1,2,3]
1 WHO Scientific Advisory Group for the Origins of Novel Pathogens (SAGO). Independent assessment of the origins of SARS-CoV-2. World Health Organization. 2025. URL: https://cdn.who.int/media/docs/default-source/documents/epp/sago/independent-assessment-of-the-origins-of-sars-cov-2-by-sago.pdf?sfvrsn=b0f90ad4_4, zuletzt abgerufen am: 05.09.2025
2 Schneider L. WHO report suggests likely zoonotic origin of Covid-19, doesn't rule out lab leak. JAMA. 2025; 334(8):661-662. doi: 10.1001/jama.2025.10976
3 Crits-Christoph A. et al. Genetic tracing of market wildlife and viruses at the epicenter of the COVID-19 pandemic. Cell. 2025; 187(19):5468-5482.e11. doi: 10.1016/j.cell.2024.08.010
10.09.2025
STIKO-Empfehlung für Impfung gegen Chikungunya
Die Ständige Impfkommission am Robert-Koch-Institut (STIKO) hat in Zusammenarbeit mit der Deutschen Gesellschaft für Tropenmedizin, Reisemedizin und Globale Gesundheit e.V. (DTG) eine Empfehlung zur Chikungunya-Impfung erarbeitet [1]. Ziel der Empfehlung ist die Verhinderung von Erkrankungen, schweren Krankheitsverläufen und Tod durch eine Chikungunya-Infektion.
Chikungunya-Fälle wurden in Deutschland bisher nur bei Reiserückkehrern detektiert. Das allgemeine Risiko bei Reisen in Endemiegebiete wird zwar als gering eingestuft, steigt aber bei einer Reise in ein aktuelles Ausbruchsgebiet. Derzeit kommen verhältnismäßig viele infizierte Reisende aus Ländern mit Chikungunya-Ausbrüchen nach Deutschland zurück: im Vergleich zu normalerweise 2-5 Chikungunya-Fällen pro Monat in den Vorjahren, wurden dem RKI zwischen April und Juni 2025 bereits insgesamt 75 Fälle gemeldet, fast ausschließlich erworben auf Mauritius (33 %), La Réunion (29 %) und Sri Lanka (9 %).
Die STIKO empfiehlt die Reiseimpfung gegen Chikungunya daher allen Personen ≥ 12 Jahren,
- die in ein Gebiet reisen, für das ein aktuelles Chikungunya-Ausbruchsgeschehen bekannt ist,
- die einen längeren Aufenthalt (> 4 Wochen) oder wiederholte Kurzzeitaufenthalte in Chikungunya-Endemiegebieten planen und bei denen ein erhöhtes Risiko für eine Chronifizierung oder einen schweren Verlauf der Erkrankung besteht (z.B. Alter 60 Jahre oder eine schwere internistische Grunderkrankung).
Zudem wird Personen, die gezielte Tätigkeiten mit Chikungunya-Viren ausüben (z.B. in Forschungseinrichtungen oder Laboratorien), eine beruflich indizierte Impfung empfohlen.
Für die aktive Immunisierung ist eine Impfstoffdosis der beiden in Deutschland verfügbaren Impfstoffe VIMKUNYA oder IXCHIQ notwendig. Der Totimpfstoff VIMKUNYA ist für alle Personen ab 12 Jahren zugelassen und empfohlen, der attenuierte Lebendimpfstoff IXCHIQ soll gemäß STIKO-Empfehlung nur im Alter von 12 bis 59 Jahren eingesetzt werden. Innerhalb dieser Altersbereiche gelten beide Impfstoffe allgemein als gleichwertig.
Aufgrund eines unterschiedlichen Wirksamkeits- und Sicherheitsprofils kann bei häufigeren Reisen oder längeren beruflichen Einsätzen in Endemiegebieten der Lebendimpfstoff IXCHIQ bevorzugt werden, sofern keine Kontraindikationen wie eine bestehende oder geplante Schwangerschaft oder Immundefizienz bestehen. Da VIMKUNYA in den Zulassungsstudien ein günstigeres Sicherheitsprofil aufwies, ist für einmalige Reisen oder bei erhöhter Relevanz möglicher Nebenwirkungen eher der Totimpfstoff VIMKUNYA zu erwägen.
Zum Zeitpunkt der Veröffentlichung der STIKO-Empfehlung (10.07.2025) war die Anwendung von IXCHIQ bei Personen ≥ 65 Jahren vorübergehend kontraindiziert, nachdem im Frühjahr 2025 eine Reihe von Fallberichten über schwerwiegende Nebenwirkungen in dieser Altersgruppe bekannt geworden waren [2]. Nach Abschluss der Überprüfung durch den Sicherheitsausschuss (PRAC) der europäischen Arzneimittelbehörde EMA wurde am 11. Juli 2025 die Aufhebung der Altersbeschränkung bekannt gegeben, da das Nutzen-Risiko-Verhältnis des Impfstoffes weiterhin grundsätzlich positiv ist [3]. Dennoch sollte der Impfstoff bei Menschen aller Altersgruppen nur bei einem erheblichen Risiko für eine Infektion und nach sorgfältiger Nutzen-Risiko-Abwägung eingesetzt werden. Dies betrifft insbesondere vorerkrankte Personen über 65 Jahre, bei denen eine erhöhte Reaktogenität des Impfstoffes zu beobachten war und die gleichzeitig ein erhöhtes Risiko für einen schweren Verlauf der Infektion und eine Chronifizierung der Symptome aufweisen.
In Österreich wird die Impfung gegen Chikungunya Reisenden in Endemiegebiete ab dem vollendeten 12. Lebensjahr, bei epidemiologischem Risiko und exponiertem Laborpersonal empfohlen. Basierend auf dem vermutlich günstigeren Nebenwirkungsprofil, soll der Totimpfstoff VIMKUNYA bei Verfügbarkeit und entsprechender Indikation vor allem bei älteren Menschen und Personen mit Grunderkrankungen bevorzugt werden [4]. In der Schweiz sind derzeit noch keine Impfstoffe gegen Chikungunya zugelassen.
1 Kling, K. et al. Beschluss und wissenschaftliche Begründung der STIKO-Empfehlung zur Impfung gegen Chikungunya. Epid Bull 2025; 28:3-38. doi: 10.25646/13274
2 European Medicines Agency (EMA). EMA starts review of Ixchiq (live attenuated chikungunya vaccine); published online May 7, 2025. URL: https://www.ema.europa.eu/en/news/ema-starts-review-ixchiq-live-attenuated-chikungunya-vaccine [03.08.2025]
3 European Medicines Agency (EMA). Ixchiq: temporary restriction on vaccinating people 65 years and older to be lifted; published online Jul 11, 2025. URL: https://www.ema.europa.eu/en/news/ixchiq-temporary-restriction-vaccinating-people-65-years-older-be-lifted [03.08.2025]
4 Bundesministerium für Arbeit, Soziales, Gesundheit, Pflege und Konsumentenschutz. Impfempfehlung Chikungunya (Stand 25.07.2025). URL: https://www.sozialministerium.gv.at/dam/jcr:0c5032e7-6bac-4264-b538-a20fda08477e/Impfplan%20Chikungunya_25.07.2025.pdf [03.08.2025]
13.08.2025
Update zur Einstufung und Nutzenbewertung von Reserveantibiotika gemäß §35a SGB V (2025)
Version 2.2 (RKI/BfArM) aktualisiert Kriterien und Erregerliste für die indikations- und erregerspezifische Einstufung von Reserveantibiotika, die in Deutschland von der Zusatznutzenbewertung freigestellt sind.
Wichtigste Änderungen der aktuellen Version (2025)
- Datengrundlagen: Anpassung der Surveillance-Daten (ARS, Antibiotika-Resistenz-Surveillance) und Einbeziehung aktueller Resistenztrends sowie Bewertung der klinischen Relevanz.
- Kriterienanpassung: Die Einstufung eines Reserveantibiotikums erfolgt stärker erregerspezifisch und indikationsbezogen (entsprechend der Erregerliste), ohne Einschränkung auf den Schweregrad der Infektion oder die Patientengruppe.
- Indikatortabelle: Der unspezifische Begriff „potenziell schwerwiegend“ wurde gestrichen; nur klar definierte und überprüfbare Kriterien bleiben erhalten.
- Checkliste und Flowchart: Überarbeitung zur eindeutigen Zuordnung für Antragstellung und Bewertung beim G-BA.
Einstufung eines neuen Reserveantibiotikums – Kriterien des G-BA
Ein neues Antibiotikum erhält den Reserveantibiotika-Status, wenn:
- Es wirksam gegen mindestens einen relevanten multiresistenten Erreger der Erregerliste ist UND
- Nur eingeschränkte klinisch gleichwertige Therapieoptionen bestehen.
- Der Antrag muss aussagekräftige in-vitro- und in-vivo-Wirksamkeitsdaten (mindestens 10 Patienten), Leitlinienprüfung der Therapieoptionen sowie eine Bezugnahme auf die nationale Resistenzsituation enthalten.
Fazit für die Praxis
Die Einstufung erfolgt gezielt nach Erreger und Indikation. Für die Zulassung als Reserveantibiotikum sind limitierte Therapiealternativen und belegte Wirksamkeit gegen aktuelle MRE entscheidend. Die
Erregerliste dient unmittelbar als Orientierungshilfe in Klinik, Forschung und bei G BA-Anträgen.
13.08.2025
Rifaximin zusätzlich zu Antibiotika bei akuter hepatischer Enzephalopathie bei kritisch kranken Patienten mit Leberzirrhose
Die hepatische Enzephalopathie (HE) ist eine potenziell reversible Komplikation des Leberversagens, die durch unterschiedliche Grade kognitiver Beeinträchtigungen gekennzeichnet ist und häufig eine Aufnahme auf der Intensivstation (ICU) erfordert. Kritisch kranke Patienten mit Leberzirrhose erhalten auf der ICU üblicherweise Breitspektrum-Antibiotika aufgrund vermuteter Infektionen oder gemäß Krankenhausprotokoll. Bisher war unklar, ob zusätzliches Rifaximin einen synergistischen Effekt mit Breitspektrum-Antibiotika bei ICU-Patienten mit akuter manifester HE hat.
In einer doppelblinden, randomisierten kontrollierten Studie in Indien wurden zwischen Februar 2022 und April 2023 Patienten mit manifester HE auf der ICU entweder der Gruppe mit alleiniger Antibiotikatherapie (AB) oder der Gruppe mit Antibiotika plus Rifaximin (AB + R) zugeteilt. Die Baseline-Charakteristika und Schweregrad-Scores waren in beiden Gruppen (je 92 Patienten) vergleichbar. Carbapenem- und Cephalosporin-Antibiotika mit β-Lactamase-Inhibitoren waren die am häufigsten eingesetzten Antibiotika.
In der Kaplan-Meier-Analyse erreichten 44,6 % (41/92; 95-%-Konfidenzintervall [KI] 32–70,5) in der AB-Gruppe und 46,7 % (43/92; 95-%-KI 33,8–63) in der AB + R-Gruppe den primären Endpunkt einer Reduktion der HE um 2 Grade oder komplette Resolution (p = 0,84). Die Zeit bis zum Erreichen des primären Endpunkts war mit 3,65 ± 1,82 Tagen vs. 4,11 ± 2,01 Tagen vergleichbar (p = 0,27). Auch die Krankenhaussterblichkeit unterschied sich nicht signifikant zwischen den Gruppen (62 % vs. 50 %; p = 0,13). Nosokomiale Infektionen traten bei 7 % in der AB-Gruppe und 13 % in der AB + R-Gruppe auf (p = 0,21). Die Endotoxin-Spiegel wurden durch Rifaximin nicht beeinflusst.
In der Subgruppenanalyse zeigte sich bei Patienten mit dekompensierter Zirrhose unter Rifaximin eine niedrigere Krankenhaussterblichkeit (Hazard Ratio 0,39; 95-%-KI 0,2–0,76), nicht jedoch bei Patienten mit akut-auf-chronischem Leberversagen (Hazard Ratio 0,99; 95-%-KI 0,6–1,63). Dies war auf eine reduzierte Rate nosokomialer Infektionen zurückzuführen.
In der multivariaten Cox-Regressionsanalyse waren nur der Serum-Bilirubinspiegel (Hazard Ratio 0,96; 95-%-KI 0,94–0,99; p = 0,005), der Serum-Albuminspiegel (Hazard Ratio 1,68; 95-%-KI 1,12–2,5; p
= 0,01) und der SOFA-Score (Hazard Ratio 0,88; 95-%-KI 0,82–0,95; p = 0,002) bei Aufnahme prädiktiv für eine Verbesserung der HE.
Folgerung der Autoren
Die Rückbildung einer manifesten HE bei Patienten unter Antibiotikatherapie war vergleichbar mit der bei Patienten, die zusätzlich Rifaximin erhielten. Rifaximin zeigte keinen signifikanten Beitrag zur Auflösung der HE bei kritisch kranken Patienten mit Leberzirrhose auf der ICU, die bereits Lactulose und andere Antibiotika erhielten.
Kommentar der Herausgeber
Rifaximin zeigte in dieser Studie etwas überraschend außer in der Subgruppe von Patienten mit dekompensierter Leberzirrhose, keinen klinischen Nutzen. Dies könnte zum einen daran liegen, dass die Patienten insgesamt schwer krank waren und bereits bei Einschluss Infektionen hatten, und andererseits, dass Rifaximin insbesondere über die Darmmikrobiota und eine Epithelstabilisierung wirkt, aber weniger Effekte auf bakterielle Translokation und Inflammation hat.
Kulkarni, A. V. et al. Antibiotics With or Without Rifaximin for Acute Hepatic Encephalopathy in Critically Ill Patients With Cirrhosis: A Double-Blind, Randomized Controlled (ARiE) Trial. Am J
Gastroenterol. 2024; 119(5):864-874.
doi: 10.14309/ajg.0000000000002575
17.07.2025
Welt-Hepatitis-Tag 2025
Der Welt-Hepatitis-Tag am 28. Juli 2025 steht unter dem internationalen Motto „Let's break it down" und wird in Deutschland mit dem Slogan „Lass uns Klartext reden" umgesetzt. Ziel des Aktionstages ist es, Mythen und Vorurteile zu Hepatitis-Viren, Ansteckungswegen, Verläufen und Therapien aufzuklären.
Weltweit leben laut WHO-Schätzungen 254 Millionen Menschen mit Hepatitis B und 50 Millionen mit Hepatitis C – viele, ohne es zu wissen. Unbehandelt können chronische Hepatitis-Infektionen zu Zirrhose und Leberkrebs führen. Es gibt fünf verschiedene Hepatitis-Viren (A bis E), die unterschiedlich übertragen werden.
Hepatitis A und E sind meist harmlos, während Hepatitis B und C und bei Immunsupprimierten auch Hepatitis E chronisch werden können. Gegen Hepatitis A und B gibt es Impfungen, Hepatitis C ist heute fast immer heilbar. Gegen Hepatitis E sind verschiedene Impfkandidaten gegen die in Entwicklungsländern aber nicht bei uns vorherrschenden Hepatitis E Serotypen 1 und 4 zugelassen oder in Entwicklung. Hepatitis D ist zwar selten, aber besonders gefährlich. Viele Hepatitis-Infektionen verlaufen unbemerkt. Ab 35 Jahren können sich Krankenversicherte einmalig kostenfrei auf Hepatitis B und C testen lassen.
17.07.2025
Thymosin α1 zeigt keine Wirksamkeit bei Sepsis
Eine aktuelle multizentrische, doppelblinde, placebokontrollierte Phase-3-Studie untersuchte die Wirksamkeit und Sicherheit von Thymosin α1 bei der Behandlung von Sepsis. Die TESTS-Studie (The Efficacy and Safety of Thymosin α1 for Sepsis) wurde zwischen September 2016 und Dezember 2020 in 22 Zentren in China durchgeführt und umfasste 1106 erwachsene Patienten im Alter von 18 bis 85 Jahren mit einer Sepsis-Diagnose nach Sepsis-3-Kriterien.
Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder Thymosin α1 (n = 552) oder Placebo (n = 554). Die Randomisierung erfolgte stratifiziert nach Alter (< 60 und ≥ 60 Jahre) und Zentrum. Thymosin α1 wurde alle 12 Stunden über 7 Tage subkutan injiziert, sofern die Behandlung nicht aufgrund einer Entlassung von der Intensivstation, des Todes oder eines Widerrufs der Einwilligung vorzeitig beendet wurde.
Der primäre Endpunkt war die 28-Tage-Gesamtmortalität nach Randomisierung. In die modifizierte Intention-to-treat-Analyse wurden 1089 Patienten eingeschlossen, die mindestens eine Dosis der Studienmedikation erhalten hatten (Thymosin α1-Gruppe n = 542, Placebo-Gruppe n = 547). Das mediane Alter betrug 65 Jahre (Interquartilsbereich 52–73) und 68,9 % der Teilnehmer waren männlich.
Die 28-Tage-Gesamtmortalität trat bei 127 Patienten (23,4 %) in der Thymosin α1-Gruppe und bei 132 Patienten (24,1 %) in der Placebo-Gruppe auf (Hazard Ratio 0,97; 95-%-Konfidenzintervall 0,76 bis 1,24; P = 0,93 im Log-Rank-Test). Auch für die sekundären Endpunkte wie 90-Tage-Gesamtmortalität (Hazard Ratio 0,95; 95-%-KI 0,77 bis 1,17), Intensivstationsmortalität, Auftreten neuer Infektionen innerhalb von 28 Tagen und Änderung des SOFA-Scores an Tag 7 zeigten sich keine statistisch signifikanten Unterschiede zwischen den Gruppen.
In der präspezifizierten Subgruppenanalyse zeigte sich ein potenzieller differenzieller Effekt von Thymosin α1 auf den primären Endpunkt in Abhängigkeit vom Alter (< 60 Jahre: Hazard Ratio 1,67; 95-%-KI 1,04 bis 2,67; ≥ 60 Jahre: 0,81; 0,61 bis 1,09; P für Interaktion = 0,01) und Diabetes (mit Diabetes: 0,58; 0,35 bis 0,99; ohne Diabetes: 1,16; 0,87 bis 1,53; P für Interaktion = 0,04).
Insgesamt traten bei 730 Patienten (67,0 %) mindestens ein unerwünschtes Ereignis auf, ohne signifikante Unterschiede zwischen den Gruppen (Thymosin α1: 66,4 % vs. Placebo: 67,6 %; Gruppendifferenz −1,2 %; 95-%-KI −6,8 % bis 4,4 %; P = 0,70). Die häufigsten unerwünschten Ereignisse waren Anämie (10,7 %), gefolgt von Fieber (9,6 %), abdomineller Distension (5,4 %) und Gerinnungsstörungen (4,8 %). Es traten keine unerwarteten schwerwiegenden Nebenwirkungen im Zusammenhang mit Thymosin α1 auf.
In einer explorativen Analyse der Immunmarker zeigte sich nach einer Woche Behandlung in beiden Studienarmen eine ähnliche Veränderung der monozytären HLA-DR-Expression, der Lymphozytenzahl und des prozentualen Anteils regulatorischer T-Zellen. Die absolute Veränderung (Gruppendifferenz −1,7; 95-%-KI −3,0 bis −0,6; P = 0,02) und die prozentuale Veränderung (Gruppendifferenz 9 %; 2 % bis 15 %; P = 0,006) des Neutrophilen-Lymphozyten-Verhältnisses waren in der Thymosin α1-Gruppe jedoch größer als in der Placebo-Gruppe.
Folgerung der Autoren
Diese Studie fand keine eindeutigen Belege dafür, dass Thymosin α1 die 28-Tage-Gesamtmortalität bei erwachsenen Patienten mit Sepsis reduziert. Das gute Sicherheitsprofil des Medikaments bei der Behandlung von Sepsis wurde jedoch bestätigt. Zusätzlich könnte Thymosin α1 möglicherweise vorteilhafte Effekte bei Patienten im Alter von 60 Jahren und älter sowie bei Patienten mit chronischen Erkrankungen haben. Zukünftige Forschung sollte sich auf diese Patienten konzentrieren, um potenzielle therapeutische Vorteile zu klären.
Wu, J. et al. The efficacy and safety of thymosin α1 for sepsis (TESTS): multicentre, double blinded, randomised, placebo controlled, phase 3 trial. BMJ. 2025; 388:e082583. doi: 10.1136/bmj-2024-082583
Wu, J. et al. Corrections: The efficacy and safety of thymosin α1 for sepsis (TESTS): multicentre, double blinded, randomised, placebo controlled, phase 3 trial. BMJ. 2025; 389:r1098. doi: 10.1136/bmj.r1098
18.06.2025
INFEKTIO_Podcast | Startschwierigkeiten – Die Neugeborenensepsis
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuellen Folge ist Prof. Dr. Christian Gille aus Heidelberg zu Gast. Gemeinsam widmen sie sich dem Thema Neugeborenensepsis.
18.06.2025
Rückkehr eliminierter Infektionskrankheiten bei sinkenden Impfraten in den USA
Die flächendeckende Durchführung von Routineimpfungen im Kindesalter hat in den USA zur Eliminierung verschiedener Infektionskrankheiten geführt. Allerdings sind die Impfraten in den letzten Jahren rückläufig, und es gibt politische Debatten über eine Reduzierung des Kinderimpfplans. Eine aktuelle Simulationsstudie untersuchte nun die möglichen Folgen sinkender Impfraten für die Wiederkehr von Masern, Röteln, Poliomyelitis und Diphtherie.
Die Modellierung erfolgte für alle 50 US-Bundesstaaten und den District of Columbia über einen Zeitraum von 25 Jahren. Dabei wurden Daten zur Demografie, Bevölkerungsimmunität und zum Importrisiko für die jeweiligen Infektionskrankheiten berücksichtigt. Mit den aktuellen bundesstaatlichen Impfraten prognostiziert das Modell, dass Masern in 83 % der Simulationen nach durchschnittlich 20,9 Jahren wieder endemisch werden könnten. Dabei würden über 25 Jahre schätzungsweise 851.300 Masernfälle (95-%-Konfidenzintervall [KI] 381.300 bis 1,3 Millionen Fälle) auftreten. Bei einem 10-prozentigen Rückgang der Masern-Mumps-Röteln-Impfung rechnet das Modell mit 11,1 Millionen Masernfällen (95-%-KI 10,1 bis 12,1 Millionen) über 25 Jahre. Bei einer 5-prozentigen Steigerung der Impfrate würden dagegen nur etwa 5800 Fälle (95-%-KI 3100 bis 19.400) erwartet.
Bei einem Rückgang der Routineimpfungen um 50 % prognostiziert das Modell über 25 Jahre 51,2 Millionen Masernfälle (95-%-KI 49,7 bis 52,5 Millionen), 9,9 Millionen Rötelnfälle (95-%-KI 6,4 bis 13,0 Millionen), 4,3 Millionen Poliomyelitisfälle (95-%-KI 4 Fälle bis 21,5 Millionen) und 197 Diphtheriefälle (95-%-KI 1 bis 1000). Dies würde zu schwerwiegenden Komplikationen führen: 51.200 Fälle (95-%-KI 49.600 bis 52.600) mit neurologischen Folgeerscheinungen nach Masern, 10.700 Fälle (95-%-KI 6700 bis 14.600) mit kongenitalem Rötelnsyndrom, 5400 Fälle (95-%-KI 0 bis 26.300) mit paralytischer Poliomyelitis sowie insgesamt 10,3 Millionen Krankenhauseinweisungen (95-%-KI 9,9 bis 10,5 Millionen) und 159.200 Todesfälle (95-%-KI 151.200 bis 164.700).
In diesem Szenario würden Masern nach durchschnittlich 4,9 Jahren (95-%-KI 4,3 bis 5,6 Jahre) endemisch werden, Röteln nach 18,1 Jahren (95-%-KI 17,0 bis 19,6 Jahre). Poliomyelitis würde in etwa der Hälfte der Simulationen (56 %) nach durchschnittlich 19,6 Jahren (95-%-KI 14,0 bis 24,7 Jahre) endemisch. Die Wahrscheinlichkeit einer endemischen Übertragung von Diphtherie wäre selbst bei diesem starken Impfrückgang gering.
Die Modellierung zeigte deutliche regionale Unterschiede beim Risiko für Ausbrüche und die Rückkehr zu einer endemischen Übertragung. Texas wies dabei das höchste Risiko für Masernausbrüche auf.
Die Ergebnisse waren robust in verschiedenen Sensitivitätsanalysen, wobei die Basisreproduktionszahl und die Importrate der Erreger den größten Einfluss auf die Resultate hatten.
Folgerung der Autoren
Basierend auf den Schätzungen dieser Modellierungsstudie würden sinkende Impfraten im Kindesalter die Häufigkeit und das Ausmaß von Ausbrüchen zuvor eliminierter impfpräventabler
Infektionskrankheiten erhöhen und schließlich zu deren Rückkehr auf ein endemisches Niveau führen. Der Zeitpunkt und die kritische Schwelle für die Rückkehr zur Endemizität unterscheiden sich dabei
erheblich je nach Krankheit, wobei Masern wahrscheinlich als erste Erkrankung zurückkehren und dies möglicherweise bereits unter den aktuellen Impfraten geschehen könnte, wenn keine Verbesserung der
Impfabdeckung und der Reaktion des öffentlichen Gesundheitswesens erfolgt. Diese Erkenntnisse unterstreichen die Notwendigkeit, Routineimpfungen im Kindesalter mit hoher Durchimpfungsrate
fortzusetzen, um ein Wiederauftreten impfpräventabler Infektionskrankheiten in den USA zu verhindern.
Kommentar der Herausgeber
Diese Modellrechnungen basieren auf konstanten Impfraten. Dagegen ist jedoch ebenso denkbar, dass ein gehäuftes Wiederauftreten dieser Erkrankungen die Gefährlichkeit dieser in weiten Kreisen der
Bevölkerung vergessenen und verharmlosten Infektionen wieder in Erinnerung rufen und Impfraten auch konsekutiv ansteigen könnten. Betroffen wären insbesondere Kinder, Alte und Immunsupprimierte.
Allerdings sollten alle Anstrengungen unternommen werden, dass es gar nicht erst zu einem weiteren Abfall der Impfraten kommt.
Kiang, M. V. et al. Modeling Reemergence of Vaccine-Eliminated Infectious Diseases Under Declining Vaccination in the US. JAMA. 2025; published online April 24, 2025.
doi: 10.1001/jama.2025.6495
21.05.2025
WHO aktualisiert Liste der prioritären antibiotikaresistenten Erreger für 2024
Die Antibiotikaresistenz stellt eine der größten Herausforderungen für die globale Gesundheit dar. Im Jahr 2019 wurden schätzungsweise 4,95 Millionen Todesfälle mit antibiotikaresistenten Erregern in Verbindung gebracht, wobei Länder mit niedrigem und mittlerem Einkommen überproportional betroffen waren.
Die Weltgesundheitsorganisation (WHO) hat nun ihre „Bacterial Priority Pathogens List" (BPPL) für 2024 aktualisiert. Diese Liste, die erstmals 2017 veröffentlicht wurde, dient als Orientierungshilfe für Investitionen in Forschung und Entwicklung sowie als Grundlage für Maßnahmen zur Überwachung und Kontrolle von Antibiotikaresistenzen. Die aktualisierte Liste umfasst 15 Familien antibiotikaresistenter Erreger, die in die Kategorien „kritisch", „hoch" und „mittel" eingestuft wurden.
In der Kategorie der kritischen Priorität finden sich Gram-negative Bakterien, die gegen Reserveantibiotika resistent sind. Dazu gehören Acinetobacter baumannii und verschiedene Erreger aus der Ordnung der Enterobacterales sowie Rifampicin-resistente Mycobacterium tuberculosis. Diese Einstufung basiert auf ihrer Fähigkeit, Resistenzgene zu übertragen, der Schwere der von ihnen verursachten Infektionen und Erkrankungen sowie ihrer erheblichen globalen Krankheitslast, insbesondere in Ländern mit niedrigem und mittlerem Einkommen.

Die Aufnahme von Salmonella und Shigella in die Kategorie hoher Priorität spiegelt ihre zunehmende Resistenz gegen bestehende Behandlungen und die hohe Infektionslast wider, die mit diesen Erregern verbunden ist.
Zu den weiteren Erregern mit hoher Priorität gehören antibiotikaresistente Pseudomonas aeruginosa und Staphylococcus aureus aufgrund ihrer globalen Bedrohung, insbesondere im Gesundheitswesen. Auch Neisseria gonorrhoeae wurde in diese Kategorie aufgenommen, da multiresistente Stämme die Behandlungsoptionen einschränken. Ein weiterer Erreger von besonderer Bedeutung für die öffentliche Gesundheit ist der antibiotikaresistente Enterococcus faecium, der aufgrund seiner Fähigkeit, Resistenzelemente über das gesamte One-Health-Spektrum zu übertragen, als besonders wichtig eingestuft wird.

In der Kategorie mittlerer Priorität listet die BPPL 2024 Streptokokken der Gruppen A und B, Streptococcus pneumoniae und Haemophilus influenzae auf. Dies weist auf einen dringenden Handlungsbedarf hin, insbesondere im Hinblick auf vulnerable Bevölkerungsgruppen in ressourcenarmen Umgebungen.

Die Bekämpfung der Antibiotikaresistenz im Gesundheitssektor erfordert globale Anstrengungen in mehreren Schlüsselbereichen: robuste Maßnahmen zur Infektionsprävention und -kontrolle, die Sicherstellung eines gerechten Zugangs zu Diagnostik und Behandlung, eine aufmerksame Überwachung zur Erkennung neuer Resistenzentwicklungen sowie erhebliche Investitionen in Forschung und Entwicklung für neue Medikamente, Diagnostika und Präventionstools.
Mehr Informationen zur WHO Bacterial Priority Pathogens List, 2024
21.05.2025
INFEKTIO_Podcast | Vom Furunkel zur Fasziitis – Haut- und Weichgewebeinfektionen
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für Patienten
auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuellen Folge ist Prof. Dr. Christian Eckmann aus Hann Münden zu Gast. Gemeinsam widmen sie sich dem Thema Haut- und Weichgewebeinfektionen.
21.05.2025
Gepotidacin zur Behandlung unkomplizierter Harnwegsinfekte in den USA zugelassen
Die US-amerikanische Food and Drug Administration (FDA) hat unter dem Namen Blujepa das neue orale Antibiotikum Gepotidacin zur Behandlung von Frauen und Mädchen ab 12 Jahren mit unkomplizierten Harnwegsinfektionen, verursacht durch Escherichia coli, Klebsiella pneumoniae, Citrobacter freundii complex, Staphylococcus saprophyticus oder Enterococcus faecalis, zugelassen [1].
Schätzungen zufolge sind 50-60 % der erwachsenen Frauen in ihrem Leben mindestens einmal von einer Harnwegsinfektion betroffen, die in den allermeisten Fällen durch E. coli verursacht wird [2]. Wenn im Harntrakt keine relevanten, funktionellen oder anatomischen Anomalien, keine relevanten Nierenfunktionsstörungen und keine relevanten Vor- bzw. Begleiterkrankungen vorliegen, die eine Harnwegsinfektion bzw. gravierende Komplikationen begünstigen, wird die Harnwegsinfektion als unkompliziert eingestuft [3].
Gepotidacin ist ein neuartiges Antibiotikum mit Triazaacenaphthylen-Struktur und damit der erste Vertreter einer neuen Wirkstoffklasse. Gepotidacin wirkt bakterizid, indem es sowohl die DNA-Gyrase als auch die Topoisomerase IV inhibiert und damit die bakterielle DNA-Replikation hemmt [1,4]. Im Unterschied zu den Fluorchinolonen interagiert Gepotidacin mit anderen Bindungsstellen auf den beiden Typ-II-Topoisomerasen und induziert dabei enzymvermittelte Einzelstrang- statt Doppelstrang-DNA-Brüche [4].
Blujepa enthält 750 mg Gepotidacin (als Gepotidacin-Mesylat) pro Tablette, die Einnahme erfolgt zweimal täglich über 5 Tage zu den Mahlzeiten, um das Risiko für gastrointestinale Beschwerden zu minimieren [1]. Eine Dosisanpassung bei leichter bis moderater Nieren- oder Leberfunktionseinschränkung ist nicht erforderlich.
Die Wirksamkeit und Sicherheit von Gepotidacin wurden in zwei randomisierten, doppelblinden Phase-III-Nichtunterlegenheitsstudien (EAGLE-2 und EAGLE-3) an nicht-schwangeren Frauen und Mädchen ab
12 Jahren (mind. 40 kg Körpergewicht) untersucht [5]. Eingeschlossen wurden Patientinnen mit mindestens zwei Symptomen eines unkomplizierten Harnwegsinfekts (Dysurie, Häufigkeit, Dringlichkeit,
Unterleibsschmerzen) sowie dem Nachweis von Nitrit im Urin und/oder dem Vorliegen einer Pyurie. Die Patientinnen erhielten über die Dauer von fünf Tagen entweder zweimal täglich 750 mg Gepotidacin
als Tablette oder zweimal täglich 100 mg Nitrofurantoin als Kapsel (25 % Nitrofurantoin makrokristallin, 75 % Nitrofurantoin-Monohydrat), sowie zusätzlich die jeweils andere Darreichungsform als
Placebo zur besseren Verblindung. Der kombinierte primäre Endpunkt galt als erreicht, wenn an Tag 10 (bis 13) sowohl eine klinische Heilung der Symptome als auch eine mikrobiologische
Eradikation
(< 10³ koloniebildende Einheiten (KBE)/ml im Urin) erreicht wurde.
In der Zwischenanalyse wurden Patientinnen berücksichtigt, bei denen vollständige mikrobiologische Daten verfügbar waren und ein Nitrofurantoin-sensibler Keim zu Studienbeginn nachgewiesen werden konnte. In EAGLE-2 erreichten unter diesen Bedingungen 50,6 % (162/320) der Patientinnen unter Gepotidacin und 47,0 % (135/287) der Patientinnen unter Nitrofurantoin den kombinierten primären Endpunkt (adjustierte Differenz 4,3 %; 95 %-Konfidenzintervall (KI) –3,6 bis 12,1). In EAGLE-3 führte die Behandlung mit Gepotidacin bei 58,5 % (162/277) und mit Nitrofurantoin bei 43,6 % (115/264) der Patientinnen zum Therapieerfolg (adjustierte Differenz 14,6 %; 95 %-KI 6,4 bis 22,8). Damit war Gepotidacin in beiden Studien einer Therapie mit Nitrofurantoin nicht unterlegen, in EAGLE-3 war Gepotidacin der Behandlung mit Nitrofurantoin zudem überlegen. Gepotidacin zeigte dabei auch numerisch eine bessere Wirksamkeit gegenüber arzneimittelresistenten E. coli-Isolaten, die Extended Spectrum β-Lactamasen (ESBL) exprimierten bzw. fluorchinolon- oder multiresistent waren.
Die häufigsten unerwünschten Nebenwirkungen von Gepotidacin sind Diarrhö (16 %), Übelkeit (9 %) und Bauchschmerzen (4 %) [1]. Außerdem kann unter Gepotidacin eine dosisabhängige Verlängerung der QTc-Zeit auftreten.
Gepotidacin ist nach langer Zeit der erste Vertreter einer neuen Antibiotikaklasse. Der Wirkmechanismus ist ähnlich den Fluorchinolonen, wobei Gepotidacin auch bei einer Fluorchinolon-Resistenz noch wirksam sein kann. Angesichts des Auftretens von Antibiotikaresistenzen könnte Gepotidacin eine zusätzliche orale Therapieoption für die Behandlung unkomplizierter Harnwegsinfekte sein, die durch multiresistente E. coli verursacht werden. Ob sich die aufgrund des dualen Wirkmechanismus bestehende Hoffnung auf ein geringeres Potential für eine Resistenzentwicklung erfüllt, wird sich erst im Zuge der breiten Anwendung zeigen. Derzeit wird außerdem die Wirksamkeit von Gepotidacin zur Behandlung von urogenitalen Infektionen durch Neisseria gonorrhoeae, einschließlich multiresistenter Stämme, untersucht.
1 U.S. Food & Drug Administration (FDA). Blujepa (Gepotidacin): Full Prescribing Information. Mar 2025. URL: https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/218230s000lbl.pdf [13.04.2025]
2 Medina M., Castillo-Pino E. An introduction to the epidemiology and burden of urinary tract infections. Ther Adv Urol. 2019; 11:3-7. doi: 10.1177/1756287219832172
3 Deutsche Gesellschaft für Urologie e. V. S3 Leitlinie: Epidemiologie, Diagnostik, Therapie, Prävention und Management unkomplizierter, bakterieller, ambulant erworbener Harnwegsinfektionen bei Erwachsenen – Aktualisierung 2024. Langversion, 3.0, AWMF Registernummer: 043/044. URL: https://register.awmf.org/de/leitlinien/detail/043-044 [13.04.2025]
4 Watkins R. et al. Gepotidacin: a novel, oral, ‘first-in-class’ triazaacenaphthylene antibiotic for the treatment of uncomplicated urinary tract infections and urogenital gonorrhoea. J Antimicrob Chemother. 2023; 78(5):1137-1142. doi: 10.1093/jac/dkad060
5 Wagenlehner F. et al. Oral gepotidacin versus nitrofurantoin in patients with uncomplicated urinary tract infection (EAGLE-2 and EAGLE-3): two randomised, controlled, double-blind, double-dummy, phase 3, non-inferiority trials. The Lancet. 2024; 403(10428):741-755. doi: 10.1016/S0140-6736(23)02196-7
25.04.2025
Weltweiter Tag der Händehygiene 2025: Es könnten Handschuhe sein - aber es ist immer Händehygiene
Eine der zentralen Maßnahmen zur Vermeidung von Infektionen ist die gründliche Händehygiene. Um auf ihre entscheidende Rolle aufmerksam zu machen, wird jedes Jahr am 5. Mai der Internationale Tag der Händehygiene begangen. Das Datum – der 5.5. – steht symbolisch für die fünf Finger jeder Hand.
Die Weltgesundheitsorganisation (WHO) setzt dieses Jahr mit dem Welttag der Händehygiene 2025 einen wichtigen Fokus auf die korrekte Verwendung von medizinischen Handschuhen und Händehygiene im
Gesundheitswesen. Nach 17 Jahren globaler Kampagnenarbeit steht nun die Botschaft im Mittelpunkt: Medizinische Handschuhe können die Händehygiene nicht ersetzen. Sie können ebenso leicht kontaminiert
werden wie bloße Hände und bieten keinen hundertprozentigen Schutz. Nach jedem Ausziehen der Handschuhe muss eine Händedesinfektion gemäß der "5 Momente der Händehygiene" erfolgen.
Ein wichtiger Aspekt ist auch der Umweltschutz: Der übermäßige Handschuhverbrauch trägt erheblich zum medizinischen Abfall bei. Zwischen Februar und August 2020 entstanden durch 3 Milliarden
Einheiten persönlicher Schutzausrüstung täglich 591 Tonnen Abfall – größtenteils Handschuhe.
Bis 2026 soll die Überwachung der Händehygiene-Compliance als nationaler Schlüsselindikator in allen Referenzkrankenhäusern etabliert werden. Aktuell setzen dies bereits 68% der Länder um.
Die WHO ruft alle Länder und Gesundheitseinrichtungen dazu auf, optimale Händehygienepraktiken und angemessene Handschuhverwendung zu priorisieren.
Alle Details zur globalen WHO-Kampagne und den geplanten Maßnahmen finden Sie hier.
25.04.2025
Europäische Impfwoche 2025: Impfungen für alle sind menschlich möglich
Die Weltgesundheitsorganisation (WHO) richtet auch 2025 wieder die Europäische Impfwoche (European Immunization Week, EIW) in der Europäischen Region aus, um die zentrale Bedeutung von Impfungen für den Gesundheitsschutz hervorzuheben. Der Fokus der jährlich stattfindenden Kampagne, die üblicherweise in der letzten Aprilwoche und dieses Jahr vom 27. April bis zum 3. Mai durchgeführt wird, liegt dabei auf dem wichtigen Ziel, eine gleichmäßig hohe Durchimpfungsrate in allen Bevölkerungsgruppen zu erreichen und dadurch Krankheitsausbrüche effektiv zu verhindern. Unter dem Motto "Impfungen für alle sind menschlich möglich" setzt die EIW ein deutliches Zeichen für einen inklusiven Zugang zu Impfangeboten.
Die diesjährige Kampagne verfolgt einen ganzheitlichen Ansatz: Neben der breiten Streuung von Informationen über Impfstoffe wird besonders betont, dass bei den Bemühungen um einen umfassenden
Gesundheitsschutz durch Impfungen niemand zurückgelassen werden darf.
Ein besonderer Schwerpunkt liegt auf der verstärkten Zusammenarbeit mit jungen medizinischen Fachkräften und Jugendorganisationen, die sich für Impfkampagnen engagieren. Damit baut die WHO gezielt
auf bestehende und neue Partnerschaften, um die jüngere Generation als wichtige Multiplikatoren für das Thema Impfen zu gewinnen.
25.04.2025
INFEKTIO_Podcast | Volle Dosis? Antibiotika-Therapie auf der Intensivstation
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für
Patienten auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuellen Folge ist Prof. Dr. Jan Kielstein aus Berlin zu Gast. Gemeinsam widmen sie sich der Antibiotika-Therapie auf der Intensivstation.
25.04.2025
Kurze Behandlungsstrategien für Rifampin-sensible Tuberkulose: Wirksamkeit und Verträglichkeit
Tuberkulose wird in der Regel mit einem 6-monatigen Rifampin-Schema behandelt. Ob eine Strategie mit einer kürzeren Erstbehandlung zu ähnlichen Ergebnissen führen kann, ist unklar.
In der nachfolgenden Studie wurden Teilnehmer mit Rifampin-empfänglicher Lungentuberkulose nach dem Zufallsprinzip entweder einer Standardbehandlung (Rifampin und Isoniazid für 24 Wochen mit
Pyrazinamid und Ethambutol für die ersten 8 Wochen) oder einer Strategie zugewiesen, die eine Anfangsbehandlung mit einem 8-Wochen-Schema oder länger bei persistierender klinischer Erkrankung einer
verlängerten Behandlung umfasste. Es gab hier vier Strategien mit unterschiedlichen Anfangstherapien; die Nichtunterlegenheit wurde in den beiden Strategien mit vollständiger Rekrutierung analysiert,
die Anfangstherapien mit hochdosiertem Rifampin-Linezolid und Bedaquilin-Linezolid (jeweils mit Isoniazid, Pyrazinamid und Ethambutol) erhielten. Der primäre Endpunkt war eine Kombination aus Tod,
fortdauernder Behandlung oder aktiver Erkrankung in Woche 96. Die Nichtunterlegenheitsmarge betrug 12 Prozentpunkte.
Es wurden 674 Teilnehmer in der Intention-to-Treat-Population eingeschlossen. Ein Ereignis mit primärem Endpunkt trat bei 7 der 181 Teilnehmer (3,9 %) in der Standardbehandlungsgruppe auf, verglichen
mit 21 der 184 Teilnehmer (11,4 %) in der Gruppe mit einem anfänglichen Rifampin-Linezolid-Schema und 11 der 189 Teilnehmer (5,8 %) in der Gruppe mit einem anfänglichen Bedaquilin-Linzolid-Schema.
Die mittlere Gesamtbehandlungsdauer betrug 180 Tage in der Standardbehandlungsgruppe, 106 Tage in der Rifampin-Linezolid-Strategiegruppe und 85 Tage in der
Bedaquilin-Linezolid-Strategiegruppe.
Die Inzidenz von unerwünschten Ereignissen des Grades 3 oder 4, von schwerwiegenden unerwünschten Ereignissen und von Todesfällen unterschied sich nicht signifikant zwischen der
Standardbehandlungsgruppe und den beiden Strategiegruppen mit vollständiger Rekrutierung. Fast alle Rückfälle wurden als Schweregrad 1 oder 2 eingestuft; ein Teilnehmer in der
Standardbehandlungsgruppe, fünf in der Rifampin-Linezolid Gruppe und einer in der Bedaquilin-Linezolid Gruppe hatten ein unerwünschtes Ereignis des Schweregrads 3 oder 4, in Form eines Rückfall oder
einer erneuten Therapie.
In einer weiteren Übersichtsarbeit und Meta-Analyse wurden Sicherheitsdaten zu 591 Patienten, die 8 unterschiedliche Therapieregime mit Bedaquilin, Pretomanid und Linezolid (BPaL) erhalten hatten,
untersucht. Bei Patienten, die eine Tagesdosis von 1200 mg Linezolid erhalten hatten, erlitten 35 % ein unerwünschtes Ereignis des Grads 3-4 aber nur 22 % von den Patienten mit einer
Linezolid-Tagesdosis von 600 mg. Diese niedrigere und besser verträgliche Dosis wurde auch in der oben genannten randomisierten Studie verwendet.
Folgerung der Autoren
Die Studie zeigt, dass eine Strategie mit initialer Behandlung mit einem 8-wöchigen Bedaquilin-Linezolid-Regime nicht unterlegen war im Vergleich zur Standardbehandlung und mit einer kürzeren
Gesamtbehandlungsdauer verbunden war. Die Sicherheit war vergleichbar in den drei Gruppen. Dies wurde inzwischen in verschiedenen Studien gezeigt und stellt somit eine vielversprechende Alternative
zur Standardbehandlung von Tuberkulose dar.
Kommentar der Herausgeber
Es ist sehr zu begrüßen, dass immer kürzere Therapieregime für Tuberkulose getestet und für sicher und wirksam befunden werden. Leider haben diese aber auch Probleme mit unerwünschten Ereignissen,
insbesondere mit Linezolid. Daher sind Daten zur ähnlichen Wirksamkeit bei besserer Verträglichkeit einer niedrigeren Tagesdosis von 600 mg erfreulich. Gleichzeitig muss jedoch bedacht werden, dass
gerade Bedaquilin und Linezolid entscheidende Therapiepfeiler von BPaL darstellen, welches die Therapie der multiresistenten (MDR) Tuberkulose durch erheblich verkürzte Therapiedauer und gute
Wirksamkeit revolutioniert hat. Bei zunehmender und breiter Anwendung dieser beiden Medikamente für Tuberkuloseformen, die potente und gut verträgliche Alternativen in Form der bisherigen 6-monatigen
Standardtherapie aufweisen, ist ein rascher Verlust durch Resistenzentwicklung für viel dringlichere Anwendungen bei der MDR-TB zu befürchten.
Paton, N. I. et al. Treatment Strategy for Rifampin-Susceptible Tuberculosis. N Engl J Med. 2023; 388(10):873-887. doi: 10.1056/NEJMoa2212537
Hasan, T. et al. The Safety and Tolerability of Linezolid in Novel Short-Course Regimens Containing Bedaquiline, Pretomanid, and Linezolid to Treat Rifampicin-Resistant Tuberculosis: An Individual Patient Data Meta-analysis. Clin Inf Dis. 2024; 78(3):730-741. doi: 10.1093/cid/ciad653
12.03.2025
Welt-Tuberkulose-Tag 2025
Tuberkulose (TB) bleibt eine der größten Herausforderungen für die globale Gesundheit. Etwa ein Viertel der Weltbevölkerung ist mit dem Erreger Mycobacterium tuberculosis infiziert. Trotz eines Rückgangs der Neuerkrankungen in der WHO-Region Europa um durchschnittlich 4,3 % pro Jahr zwischen 2011 und 2015 stellt die Krankheit weiterhin eine erhebliche Bedrohung dar, insbesondere durch multiresistente TB. Neun der 30 am stärksten betroffenen Länder weltweit befinden sich in dieser Region.
TB wird über die Luft übertragen, wobei eine unbehandelte Person mit aktiver TB durchschnittlich 10–15 Menschen pro Jahr anstecken kann. Eine frühzeitige Diagnose und eine wirksame Behandlung sind entscheidend, um die weitere Ausbreitung zu verhindern. Die WHO verfolgt das Ziel, TB weltweit zu beenden. Der TB-Aktionsplan für die WHO-Region Europa 2016–2020 dient dabei als wichtigstes Leitinstrument, um Fortschritte im Einklang mit der globalen End-TB-Strategie 2016–2035, den Zielen für nachhaltige Entwicklung und dem europäischen Gesundheitsrahmenkonzept Health 2020 zu erzielen.
Aus diesem Grund steht der Welt-Tuberkulose-Tag 2025 unter dem Motto „Ja! Wir können die Tuberkulose beenden: Bekennen, investieren, umsetzen“. Dieses Thema betont die Notwendigkeit, sich zur
Bekämpfung der Krankheit zu verpflichten, Investitionen zu tätigen und konkrete Maßnahmen zur endgültigen Eliminierung umzusetzen.
Die WHO ruft Regierungen, Gesundheitsdienstleister und Gemeinschaften zur Zusammenarbeit auf. Notwendig sind verbesserte Früherkennung, neue Diagnosetests, Medikamente sowie verstärkte Forschung. Der
Welt-Tuberkulose-Tag soll Bewusstsein schaffen, Engagement fördern und den gemeinsamen Kampf gegen TB stärken.
Weitere Informationen zum Aktionsplan, zur Kampagne und der entsprechenden Webinar-Veranstaltung finden Sie untenstehend.
12.03.2025
Veranstaltungen der Paul-Ehrlich-Gesellschaft
Frühjahrstagung der Sektion "Antimykotische Therapie"
Was? Update Antimykotika & Diagnostik
Wo? Haus der Technik, Essen
Wann? 04.-05.04.2025
Frühjahrstagung der PEG
Was? Update Infektionsmedizin (u.a. Leitlinien, ABS, Impfung & antivirale Therapie)
Wo? Volkshaus Jena
Wann? 28.-29.04.2025
12.03.2025
INFEKTIO_Podcast | Besserwisser mit Antibiotika – Etablierung von ABS-Strukturen
Der Podcast consilium infectiorum bietet medizinischem Fachpersonal praxisrelevante Themen der klinischen Infektiologie und unterstützt dabei, evidenzbasierte Behandlungsoptionen für
Patienten auszuwählen.
In jeder Folge diskutiert Infektiologe Prof. Mathias Pletz aus Jena mit einem Expertengast einen klinischen Schwerpunkt – fundiert und dennoch in entspannter Gesprächsatmosphäre.
Besonders praktisch: Für jede Folge können Sie bis zu einem Jahr nach Veröffentlichung CME-Punkte sammeln, indem Sie die zugehörigen CME-Fragen beantworten.
In der aktuellen Folge ist Prof. Dr. Irit Nachtigall aus Berlin zu Gast. Gemeinsam widmen sie sich der Etablierung von ABS-Strukturen
12.03.2025
Zulassungsempfehlung für neuen Impfstoff gegen Chikungunya-Virus
Der Ausschuss für Humanarzneimittel (CHMP) bei der europäischen Arzneimittelbehörde EMA empfiehlt nach seiner Sitzung am 30. Januar 2025 die Zulassungserteilung für Vimkunya, einen neuen Impfstoff gegen das Chikungunya-Virus (CHIKV) [1].
Das Chikungunya-Virus ist in den (sub-)tropischen Regionen Amerikas, Afrikas und Asiens endemisch. In Frankreich wurde 2024 der einzige lokal erworbene Fall auf dem europäischen Festland berichtet (ECDC). Die Übertragung erfolgt durch den Stich einer infizierten Aedes-Mücke und führt nach einer Inkubationszeit von etwa 2-12 Tagen u.a. zu einem plötzlichen Auftreten von hohem Fieber, starken Gelenkschmerzen an Händen und Füßen sowie Hautausschlag. Auch wenn schwere Verläufe selten sind, können neurologische Komplikationen und Todesfälle auftreten. Besonders belastend ist der häufig über mehrere Monate bis sogar Jahre andauernde Genesungsprozess, in denen die Betroffenen von wiederkehrenden Gelenkschmerzen begleitet werden, die nicht selten in einer chronischen Arthritis münden können [2].
Vimkunya ist ein auf virusähnlichen Partikeln (VLP) basierender Totimpfstoff. Er enthält das rekombinant hergestellte Kapsidprotein (C) sowie die Hüllproteine E1 und E2, abgeleitet vom CHIKV-Senegal-Stamm 37997 [1]. Durch eine spontane Zusammenlagerung dieser Strukturproteine, zu den sogenannten virusähnlichen Partikeln, wird die Morphologie des Virus imitiert, was nach der Injektion zu einer effektiven Stimulation der angeborenen und adaptiven Immunantwort führt [3]. Die VLPs sind dabei nicht replikationsfähig, da sie kein virales Erbgut enthalten. Zur Verstärkung der Immunantwort sind die VLPs außerdem an Aluminiumhydroxid als Adjuvans adsorbiert. Der adjuvantierte Totimpfstoff soll als Suspension zur Injektion in einer Fertigspritze erhältlich sein und kann zur aktiven Immunisierung von Personen ab 12 Jahren eingesetzt werden.
Die Immunogenität und Sicherheit des Impfstoffes wurden in zwei randomisierten, doppelblinden und kontrollierten Phase-III-Studien untersucht [4]. Die 3254 eingeschlossenen Personen im Alter zwischen 12 und 65 Jahren erhielten entweder eine Einzeldosis Vimkunya oder eine intramuskuläre Injektion mit Placebo. Die Impfung mit Vimkunya zeigte eine Seroresponse-Rate von 98 % (1 % unter Placebo) an Tag 22, die nach etwa 6 Monaten noch bei 86 % lag. In der Studie an 413 Personen im Alter 65 Jahren und älter war die durch Vimkunya-induzierte Seroresponse-Rate an Tag 22 mit 87 % im Vergleich zu der jüngeren Altersgruppe niedriger. In beiden Studien waren die schützenden Antikörpertiter bei einem Großteil der geimpften Personen bereits an Tag 15 erreicht (Seroresponse-Rate 97 % bzw. 82 %). Die unerwünschten Wirkungen nach der Impfung waren meist mild bis moderat, am häufigsten traten Schmerzen an der Injektionsstelle, Müdigkeit, Kopfschmerzen und Myalgie auf [1,4].
Folgt die Europäische Kommission der Empfehlung des CHMP und wird Vimkunya in Europa zugelassen, wäre es bereits der zweite Impfstoff gegen das Chikungunya-Virus. Mit der Zulassung von Ixchiq im Juni 2024 bestand erstmals die Möglichkeit der aktiven Immunisierung gegen CHIKV. Ixchiq ist ein Lebendimpfstoff und enthält replikationsfähige, abgeschwächte Chikungunya-Viren, mit denen Personen ab 18 Jahren geimpft werden können. Die Impfung mit Ixchiq wird derzeit in Österreich für Reisende in Endemiegebiete empfohlen, in der Schweiz ist der Impfstoff nicht zugelassen [5,6]. Eine Impfempfehlung für Reisende aus Deutschland wird derzeit noch durch die Ständige Impfkommission am Robert Koch Institut (STIKO) erarbeitet.
1 European Medicines Agency (EMA). Vimkunya - summary of opinion. EMA/CHMP/552510/2024; published online Jan 31, 2025. URL: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-vimkunya_en.pdf
2 Auswärtiges Amt. Chikungunyafieber. Stand 20.12.2024. URL:
https://www.auswaertiges-amt.de/de/ReiseUndSicherheit/reise-gesundheit/chikungunyafieber/2562870 [08.02.2025]
3 Klausberger, M. et al. Virusähnliche Partikel – Impfstoffe, die den Eindringling imitieren. Biospektrum. 2024; 30(1):66–69. doi: 10.1007/s12268-024-2112-2
4 Richardson, J. S. et al. Safety and Immunogenicity of an Adjuvanted Chikungunya Virus (CHIKV) Virus-like Particle (VLP) Based Vaccine in Two Pivotal Phase 3 Trials, ≥12 Years of Age. Open Forum
Infect Dis. 2023; 10(Suppl 2):S1270-S1271. doi: 10.1093/ofid/ofad500.2471
5 Bundesministerium für Soziales, Gesundheit, Pflege und Konsumentenschutz (BMSGPK). Impfplan Österreich 2024/2025, Version 1.1 vom 18.12.2024; S. 132-133. URL: https://www.sozialministerium.at/dam/jcr:7b3826a7-9eb5-4835-affd-f6e6e3f62bf4/Impfplan_Österreich_2024-2025_Version_1.1.pdf
6. Bundesamt für Gesundheit (BAG). Chikungunya-Fieber. Stand 11.09.2024. URL: https://www.bag.admin.ch/bag/de/home/krankheiten/krankheiten-im-ueberblick/chikungunya.html [09.02.2025]
12.02.2025
Update | VRE vanA ST612: Weite Verbreitung in der Schweiz bestätigt
Wir berichteten bereits im März und Juni 2024 auf Basis der bisherigen Veröffentlichungen von Swissnoso über die zunehmende Ausbreitung von Vancomycin-resistentem Enterococcus faecium (VRE) vanA ST612. Nun wurde die epidemiologische Untersuchung zur interregionalen Verbreitung abgeschlossen und bestätigt eine weitreichendere Verbreitung als ursprünglich angenommen, insbesondere in den deutschsprachigen Regionen der Schweiz.
Die hohe Detektionsrate klinischer Proben (44 %) und die niedrige Quote positiver Aufnahmescreenings im Vergleich zu klinischen Fällen deuten auf eine erhebliche unbemerkte Transmission hin.
Besonders besorgniserregend ist, dass ein nosokomiales Cluster bislang nicht unter Kontrolle gebracht werden konnte.
Handlungsempfehlungen für Gesundheitseinrichtungen
Erinnerung an die entscheidende Bedeutung der folgenden kritischen Präventions- und Kontrollmaßnahmen:
✅ Striktes Screening von Risikopatienten
✅ Einhaltung von Standard- und Kontaktisolationsmaßnahmen
✅ Systematische Kontaktuntersuchungen
✅ Meldepflicht ab drei verknüpften Fällen
Um die Ausbreitung innerhalb und zwischen Gesundheitseinrichtungen weiter einzudämmen, ist die konsequente Umsetzung der Swissnoso-Empfehlungen zur Prävention und Kontrolle multiresistenter Organismen essenziell und sollte aktiv gefördert werden.
12.02.2025
Zulassungsempfehlung für neue Arzneistoffe zur COVID-19-Prävention
Am 12. Dezember 2024 hat der Ausschuss für Humanarzneimittel (CHMP) bei der europäischen Arzneimittelbehörde EMA einen weiteren COVID-19-Impfstoff (Kostaive) für Erwachsene und einen monoklonalen Antikörper (Kavigale) zur passiven Immunisierung immungeschwächter Personen ab 12 Jahren zur Zulassung empfohlen [1,2].
Kostaive (Zapomeran)
Bei Kostaive handelt es sich um einen RNA-basierten Impfstoff. Er enthält mRNA, die für das Spike-Protein von SARS-CoV-2 codiert [1]. Im Jahr 2020 erhielt der damals weltweit erste mRNA-Impfstoff eine Zulassung zum Schutz vor Infektionen mit SARS-CoV-2. Mittlerweile existieren mehrere, variantenangepasste mRNA-Impfstoffe gegen COVID-19, kürzlich erfolgte auch die Zulassung eines mRNA-Impfstoffs zum Schutz vor Erkrankungen durch das Respiratorische Synzytial-Virus. Alle bisher verfügbaren mRNA-Vakzine basieren auf nicht-replizierender mRNA, von der eine ausreichend hohe Menge enthalten sein muss, um eine adäquate Immunantwort zu induzieren [3].
Im Gegensatz dazu enthält Kostaive eine selbst-replizierende bzw. selbst-amplifizierende mRNA (sa-mRNA), die nicht nur die genetische Information für das Spike-Protein enthält, sondern auch für die virale Replikase codiert. Nach der intramuskulären Injektion der in Lipid-Nanopartikeln verpackten sa-mRNA, erfolgt in den Zellen an der Injektionsstelle neben der Translation in das Spike-Protein auch die Bildung des Enzyms Replikase. Die Replikase vervielfacht einen Teil der mRNA in der Zielzelle, wodurch auch eine erhöhte Menge an Antigen exprimiert werden kann [1,3,4]. Die Verwendung von sa-mRNA soll die im Impfstoff enthaltene mRNA-Menge bei gleichzeitig längerer Produktion größerer Antigenmengen reduzieren. Durch die Erzeugung doppelsträngiger RNA-Intermediate während der Replikation sollen zusätzliche, das angeborene Immunsystem aktivierende Effekte und damit insgesamt Vorteile hinsichtlich der Ausbildung einer langanhaltenden Immunität erzielt werden [3,4].
Die Beurteilung der Wirksamkeit von Kostaive zur Grundimmunisierung gegen COVID-19 war Gegenstand einer randomisierten, Beobachter-verblindeten Phase-IIIb-Studie an 16.107 Personen in Vietnam [5]. Dabei erhielten die Teilnehmer entweder zwei Impfdosen Kostaive (Dosis 5 µg) oder Placebo im Abstand von 28 Tagen und es wurde die Anzahl aufgetretener COVID-19-Fälle in beiden Gruppen bis Tag 92 erfasst. Die Impfstoffwirksamkeit zum Schutz vor COVID-19 jeglicher Schwere betrugt 56,6 % (95 %-Konfidenzintervall (KI): 48,7–63,3), während sie gegen schwere Verläufe 95,3 % (95 %-KI: 80,5–98,9) erreichte, insbesondere bei Infektionen mit der Delta-Variante.
In einer weiteren randomisierten, doppelblinden Phase-III-Studie wurde die Immunogenität und Sicherheit von Kostaive (Dosis 5 µg) als vierte Boosterimpfung mit dem mRNA-Impfstoff BNT162b2 (Pfizer-BioNTech, Dosis 30 µg) verglichen [3]. Die Studie umfasste 828 erwachsene Teilnehmer in Japan, die zuvor drei Dosen eines mRNA-Impfstoffs erhalten hatten. Der primäre Endpunkt zeigte, dass die durch Kostaive induzierte Immunantwort 28 Tage nach der Impfung gegen die Wuhan-Hu-1-Variante der von BNT162b2 nicht unterlegen war, gemessen an neutralisierenden Antikörper-Titern und Seroresponse-Raten. Für die Omikron-Variante BA.4/5 zeigte Kostaive eine überlegene Immunantwort.
Nach der Impfung mit Kostaive traten in den Studien am häufigsten lokale Reaktionen wie Schmerzen an der Injektionsstelle sowie systemische unerwünschte Ereignisse wie Arthralgie, Myalgie, Kopfschmerzen, Schwindel, Müdigkeit, Schüttelfrost und Fieber auf. Die unerwünschten Ereignisse waren überwiegende mild bis moderat und vorübergehend [3,5].
Kavigale (Sipavibart)
Kavigale enthält den Wirkstoff Sipavibart, einen rekombinanten humanen monoklonalen IgG1-Antikörper mit der Fähigkeit, an die Rezeptor-Bindungsdomäne des Spike-Proteins von SARS-CoV-2 zu binden. Auf diesem Weg wird die Interaktion des Spike-Proteins mit dem Wirtsrezeptor ACE-2 gehemmt und das Eindringen des Virus verhindert. Eine so erreichte passive Immunisierung gegen SARS-CoV-2 soll Sipavibart immungeschwächte Personen ab 12 Jahren mit eingeschränkter Impfantwort vor einer symptomatischen COVID-19-Erkrankung schützen [2].
In den vergangenen Jahren der Pandemie wurden bereits monoklonale Antikörper und -kombinationen für die Therapie und die Prä-Expositionsprophylaxe (PrEP) von COVID-19 eingesetzt. Aktuellen Empfehlungen zufolge soll eine PrEP mit den SARS-CoV-2-neutralisierenden monoklonalen Antikörper Tixagevimab/Cilgavimab (Evusheld) nur noch in begründeten Einzelfällen in Betracht gezogen werden [6]. Der Grund dafür ist die fehlende (Tixagevimab) bzw. reduzierte (Cilgavimab) Neutralisationskapazität in in-vitro-Untersuchungen für mehrere derzeit zirkulierende Omikron-Sublinien infolge von Mutationen im Spike-Protein. Andere, zu Beginn der Pandemie eingesetzte, Antikörper sind auf Grund der fehlenden Wirksamkeit gar nicht mehr verfügbar oder werden nicht mehr empfohlen (z.B. Casirivimab / Imdevimab).
Da für eine wirksame PrEP bis dato Wirkstoffe mit breiter neutralisierender Aktivität gegenüber den vorherrschenden Virus-Varianten fehlten, wurde der lang wirkende monoklonale Antikörper Sipavibart entwickelt [7]. Sipavibart wurde aus den B-Zellen von rekonvaleszenten Patienten nach einer SARS-CoV-2-Infektion gewonnen. In der noch laufenden, randomisierten, doppelblinden Phase-III-Studie SUPERNOVA wurde die Wirksamkeit von Sipavibart bei 3.335 immungeschwächten Patienten ab 12 Jahren gegenüber Placebo und Tixagevimab/Cilgavimab untersucht. Die Patienten erhielten zwei intramuskuläre Injektionen mit je 300 mg Sipavibart oder der Vergleichssubstanz im Abstand von 6 Monaten. Ersten Ergebnissen zufolge reduzierte Sipavibart im Vergleich zu Tixagevimab/Cilgavimab oder Placebo signifikant die Inzidenz von symptomatischen SARS-CoV-2-Infektionen, unabhängig von der Virus-Variante (Beginn der Studie Dezember 2022). Der Effekt von Sipavibart war erwartungsgemäß größer gegen die Virus-Varianten ohne F456L-Mutation, in deren Anwesenheit bereits eine fehlende Neutralisationswirkung von Sipavibart bekannt ist.
Kavigale ist allgemein gut verträglich, die häufigsten Nebenwirkungen sind Reaktionen an der Injektions- bzw. Infusionsstelle [2].
Beide zur Zulassung empfohlenen Arzneimittel können zur Verbesserung und Erweiterung der Möglichkeiten der aktiven und passiven Immunisierung gegen COVID-19 beitragen. Die Zulassung und Genehmigung des Inverkehrbringens durch die Europäische Kommission ist noch ausstehend. Es bleibt abzuwarten, welchen Stellenwert sowohl Kostaive als auch Kavigale in der Prophylaxe von SARS-CoV-2-Infektionen durch aktuell (und zukünftig) zirkulierende Virus-Varianten einnehmen werden.
1 European Medicines Agency (EMA). Kostaive (Zapomeran) – summary of opinion. EMA/392588/2024; published online Dec 13, 2024. URL: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-kostaive_en.pdf [05.01.2025]
2 European Medicines Agency (EMA). Kavigale (Sipavibart) – summary of opinion. EMA/CHMP/562567/2024; published online Dec 13, 2024. URL: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-kavigale_en.pdf [05.01.2025]
3 Oda, Y. et al. Immunogenicity and safety of a booster dose of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2 mRNA COVID-19 vaccine: a double-blind, multicentre, randomised, controlled, phase 3, non-inferiority trial. Lancet Infect Dis. 2024; 24(4):351–60. doi: 10.1016/S1473-3099(23)00650-3
4 Schmidt, C. et al. Self-Amplifying RNA Vaccine Candidates: Alternative Platforms for mRNA Vaccine Development. Pathogens. 2023; 12(1):138. doi: 10.3390/pathogens12010138
5 Ho, N.T. et al. Safety, immunogenicity and efficacy of the self-amplifying mRNA ARCT-154 COVID-19 vaccine: pooled phase 1, 2, 3a and 3b randomized, controlled trials. Nat Commun. 2024; 15:4081. doi: 10.1038/s41467-024-47905-1
6 RKI (Robert Koch-Institut). COVID-19-Therapieempfehlungen: Interaktive Orientierungshilfe für Ärztinnen und Ärzte. Stand: 10.10.2024. URL: https://multimedia.gsb.bund.de/RKI/covid-19/dgiin_pv_covid-19-therapie/#/ [08.01.2025]
7 AstraZeneca. SUPERNOVA Phase III trial of sipavibart long-acting antibody met primary endpoints in preventing COVID-19 in immunocompromised patient population. Press Announcement; published online
May 16, 2024. URL: https://www.astrazeneca.com/media-centre/press-releases/2024/supernova-trial-met-covid-19-prevention-endpoint.html [08.01.2025]
15.01.2025
Neues Reserveantibiotikum Meropenem / Vaborbactam verfügbar
Mehr als fünf Jahre nach der Zulassung durch die europäische Arzneimittelbehörde EMA wurde Ende 2024 das Reserveantibiotikum Vaborem (vormals Vabomere) in Deutschland eingeführt. Es kombiniert Meropenem mit dem neuartigen β-Lactamase-Inhibitor (BLI) Vaborbactam zur Behandlung schwerer Infektionen wie u.a. komplizierten Harnwegsinfektionen (cUTI), komplizierten intraabdominellen Infektionen (cIAI) oder nosokomial erworbenen Pneumonien (HAP) [1].
Carbapenem-resistente Enterobacterales (CRE) zählen laut der Weltgesundheitsorganisation (WHO) aufgrund ihrer Verbreitung, Resistenz und der begrenzten Behandlungsmöglichkeiten weiterhin zu den kritischsten Gram-negativen Erregern. Eine Strategie, die durch β-Lactamase-Enzyme vermittelte Resistenz zu überwinden, ist die Kombination von β-Lactam-Antibiotika mit geeigneten BLI.
Vaborbactam, ein Nicht-β-Lactam BLI mit zyklischer Boronsäure-Struktur, hemmt insbesondere β-Lactamasen der Ambler-Klasse A (inkl. Klebsiella pneumoniae Carbapenemase, KPC) sowie AmpC [1,2]. Die Boronsäure-Struktur ist entscheidend für die Hemmung der Enzymaktivität der genannten β-Lactamasen, bei der es zur Ausbildung eines kovalenten Adduktes mit dem hydrolysestabilen Vaborbactam kommt. Gegen β-Lactamasen der Klasse B (Metallo-β-Lactamasen) und Carbapenemasen der Klasse D (z.B. OXA-48) besitzt Vaborbactam keine Aktivität.
Die Zulassung von Vaborem basiert hauptsächlich auf einer randomisierten, kontrollierten und doppelblinden Phase-III-Studie an 545 erwachsenen Patienten mit cUTI oder akuter Pyelonephritis [3]. Die Patienten wurden entweder mit Meropenem/Vaborbactam (2 g/2 g alle 8 h) oder Piperacillin/Tazobactam (4 g/0,5 g alle 8 h) über 10 Tage behandelt, mit der Möglichkeit einer Oralisierung auf Levofloxacin (500 mg alle 24 h). Gemessen am Gesamterfolg nach Ende der intravenösen Behandlung, ein kombinierter primärer Endpunkt aus klinischer Heilung und mikrobiologischer Eradikation, war Meropenem/Vaborbactam einer Therapie mit Piperacillin/Tazobactam nicht unterlegen (98,4 % vs. 94,0 %, Differenz: 4,5 % (95 %-Konfidenzintervall 0,7 – 9,1)). Informationen zur Wirksamkeit gegen CRE konnten in dieser Studie allerdings nicht geliefert werden, da fast keine Meropenem-resistenten Erreger behandelt wurden und so die Angemessenheit der Vaborbactam-Dosis nicht belegt werden konnte [4].
Eine weitere Phase-III-Studie an 77 Patienten mit schweren Infektionen (darunter cUTI, cIAI, HAP) verglich Meropenem/Vaborbactam mit der bestverfügbaren Therapie (Polymyxine, Carbapeneme, Aminoglykoside, Tigecyclin oder Ceftazidim-Avibactam) [5]. Bei den 47 Patienten mit CRE wurden höhere Heilungsraten und eine geringere Sterblichkeit unter Meropenem/Vaborbactam beobachtet. Die Studie diente jedoch nur einem deskriptiven Wirksamkeitsvergleich [4].
Die häufigsten unerwünschten Ereignisse unter der Behandlung mit Meropenem/Vaborbactam waren Kopfschmerz, Diarrhö, Phlebitis an der Infusionsstelle und Übelkeit [1].
Die Kombination des neuen BLI Vaborbactam mit Meropenem kann das Problem der Carbapenem-Resistenz nicht vollständig lösen, stellt jedoch eine willkommene Therapieoption für die Behandlung von
Infektionen durch CRE dar, insbesondere in Anwesenheit von KPC. β-Lactamasen der Klasse B und Carbapenemasen der Klasse D als weitere wichtige Ursachen
der Carbapenem-Resistenz werden allerdings nicht adressiert.
Der Stellenwert der neuen Kombination in der Praxis, insbesondere im Vergleich zu anderen Substanzen mit Wirksamkeit bei CRE (z.B. Imipenem/Cilastatin/Relebactam, Ceftazidim/Avibactam), ist vor dem
Hintergrund der begrenzten klinischen Daten noch offen. In Österreich ist Vaborem bereits seit 2019, in der Schweiz seit 2021 verfügbar.
1 Fachinformation Vaborem (Meropenem/Vaborbactam). Berlin-Chemie. Stand 07/2023. URL: https://www.medical-hub.berlin-chemie.de/sites/g/files/fugoka451/files/products/documents/147730_vaborem_fi-0723.pdf [11.01.2025]
2 Marino, A. et al. Ceftazidime/Avibactam and Meropenem/Vaborbactam for the Management of Enterobacterales Infections: A Narrative Review, Clinical Considerations, and Expert Opinion. Antibiotics.
2023; 12(10):1521. doi: 10.3390/antibiotics12101521
3 Kaye, K. S. et al. Effect of Meropenem-Vaborbactam vs Piperacillin-Tazobactam on Clinical Cure or Improvement and Microbial Eradication in Complicated Urinary Tract Infection: The TANGO I
Randomized Clinical Trial. JAMA. 2018; 319(8):788-799. doi: 10.1001/jama.2018.0438
4 European Medicines Agency (EMA). Assessment report Vabomere (Meropenem/Vaborbactam). Stand 20.09.2018. URL: https://www.ema.europa.eu/en/documents/assessment-report/vabomere-epar-public-assessment-report_en.pdf [11.01.2025]
5 Wunderink, R. G. et al. Effect and Safety of Meropenem-Vaborbactam versus Best-Available Therapy in Patients with Carbapenem-Resistant Enterobacteriaceae Infections: The TANGO II Randomized
Clinical Trial. Infect Dis Ther. 2018; 7(4):439-455. doi: 10.1007/s40121-018-0214-1
15.01.2025
►COVID-19 Kompendium (4., erw. u. akt. Auflage)
►COVID-19 Erkrankungen weltweit
Website der Johns Hopkins University
►COVID-19 Erkrankungen in Deutschland (Landkreise)
Jetzt für den INFEKTIO letter anmelden und regelmäßig per E-Mail Wissenswertes aus Mikrobiologie, Arznei-mittelforschung,Therapie uvm. erhalten.
Informationen für Ärzte und Apotheker zur rationalen Infektionstherapie
Die Zeitschrift für Infektionstherapie (bis 2015: "Zeitschrift für Chemotherapie") erscheint im Jahr 2026 im 47. Jahrgang. Herausgeber und Redaktion sind bemüht, Sie kontinuierlich und aktuell über wichtige Entwicklungen im Bereich der Infektionstherapie zu informieren.

